
Tue, May 19, 2026


Volume 13, Issue 3 (7-2025)
J. Pediatr. Rev 2025, 13(3): 269-276 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Karimi Rouzbahani A, Tahmasebi Ghorabi S, Kargarfard Jahromi M, Emami S, Mahmoodi M, Molavi M A. Prevalence and Correlation of Pulmonary Hypertension in Patients With Sickle Cell Disease: A Descriptive Study. J. Pediatr. Rev 2025; 13 (3) :269-276
URL: http://jpr.mazums.ac.ir/article-1-621-en.html
URL: http://jpr.mazums.ac.ir/article-1-621-en.html
Arian Karimi Rouzbahani1 

 , Samaneh Tahmasebi Ghorabi2 , Mohammadreza Kargarfard Jahromi3 , Salahaddin Emami4 , Masoumeh Mahmoodi5 , Mohammad Ali Molavi *6
, Samaneh Tahmasebi Ghorabi2 , Mohammadreza Kargarfard Jahromi3 , Salahaddin Emami4 , Masoumeh Mahmoodi5 , Mohammad Ali Molavi *6
, Samaneh Tahmasebi Ghorabi2 , Mohammadreza Kargarfard Jahromi3 , Salahaddin Emami4 , Masoumeh Mahmoodi5 , Mohammad Ali Molavi *6
1- Razi Herbal Medicines Research Center, Lorestan University of Medical Sciences, Khorramabad, Iran. & Surgery Education and Researching Network (SERGN), Universal Scientific Education and Research Network (USERN), Khorramabad, Iran.
2- Universal Scientific Education and Research Network (USERN) Office, Lorestan University of Medical Sciences, Khorramabad, Iran.
3- Department of Pediatric Cardiology, Hormozgan University of Medical Sciences, Bandar Abbas, Iran.
4- Student Research Committee, Lorestan University of Medical Sciences, Khorramabad, Iran.
5- Department of Research and Technology, Hormozgan University of Medical Sciences, Bandar Abbas, Iran.
6- Department of Pediatrics, School of Medicine, Hormozgan University of Medical Sciences, Bandar Abbas, Iran. ,shayanmolavi@yahoo.com
2- Universal Scientific Education and Research Network (USERN) Office, Lorestan University of Medical Sciences, Khorramabad, Iran.
3- Department of Pediatric Cardiology, Hormozgan University of Medical Sciences, Bandar Abbas, Iran.
4- Student Research Committee, Lorestan University of Medical Sciences, Khorramabad, Iran.
5- Department of Research and Technology, Hormozgan University of Medical Sciences, Bandar Abbas, Iran.
6- Department of Pediatrics, School of Medicine, Hormozgan University of Medical Sciences, Bandar Abbas, Iran. ,
Full-Text [PDF 413 kb]
(409 Downloads)
| Abstract (HTML) (1013 Views)
Full-Text: (256 Views)
Introduction
Sickle cell disease (SCD) is an inherited disorder caused by an aberrant type of hemoglobin S (HbS), resulting from a single-point mutation in the β-globin gene [1, 2]. This condition is notably common in areas, including India, Sub-Saharan Africa, Saudi Arabia, and South America. Individuals affected by SCD inherit two copies of the HbS gene, leading to hemolytic anemia and complications such as acute chest syndrome and vaso-occlusive pain crises (VOC). Because HbS precipitates during deoxygenation, symptoms usually manifest within the first year of life, causing erythrocytes to assume a sickle shape. This deformation disrupts blood flow, leading to hemolysis and vaso-occlusive episodes [1-3]. SCD patients are susceptible to progressive vasculopathy, which is characterized by endothelial dysfunction, systemic issues, PHT, and structural alterations, such as smooth muscle remodeling and intimal proliferation in blood vessels [1].
PHT represents a serious and potentially fatal complication of SCD, impacting the pulmonary vessels and compromising the structure and function of the right ventricle. It is characterized by a resting mean pulmonary artery pressure of more than 25 mm Hg. Notwithstanding its clinical significance, there is a lack of clarity regarding the prevalence and risk factors of PHT in individuals with SCD, and differences have been noted between studies and groups [4, 5].
PHT in SCD is associated with a range of crippling consequences, such as a worse quality of life, decreased ability to exercise, trouble with day-to-day functioning, and noticeably lower survival rates [6]. Common clinical symptoms include fatigue, peripheral edema, dyspnea, and chest pain. During the physical examination, one might observe indicators of right ventricular dysfunction, including a pronounced second heart sound, evidence of right ventricular hypertrophy, right-sided fourth heart sound, and right parasternal heave. The findings provide valuable understanding into cardiovascular functional and structural changes, aiding in diagnosis and guiding targeted management strategies [2].
The diagnosis of PHT in SCD patients typically depends on invasive right cardiac catheterization, regarded as the gold standard for assessing pulmonary artery pressure. Noninvasive methods, especially Doppler echocardiography, are employed to evaluate right ventricular dimensions and performance while estimating pulmonary artery systolic pressure (PASP). As a valuable screening tool, Doppler echocardiography helps identify people susceptible to PHT, enabling additional assessment and action [7]. Acute complications of SCD, such as vaso-occlusive crises and acute chest syndrome, can exacerbate PHT, resulting in rapid clinical decline and potentially life-threatening consequences. Thus, early identification and prompt intervention in SCD patients with PHT are essential to enhance prognosis and optimize patient outcomes [8].
SCD is one of the most common hemoglobinopathies in Iran, particularly in the southern regions. It is associated with vascular occlusive crises, severe pain, and multisystem complications; thus, studying its complications is of significant importance. However, data on the prevalence, incidence, and associated factors of PHT in the Iranian population, especially among children and adolescents under 18 years of age, are limited. This study, utilizing echocardiography and analyzing data from hospitalized patients, aimed to determine the incidence of this complication and examine its association with demographic and treatment-related factors. Therefore, the study was conducted to investigate the incidence of PHT in patients with SCD in Bandar Abbas in 2022-2023. The findings of this research could play a crucial role in early diagnosis, effective therapeutic interventions, and the development of management strategies to improve patients’ quality of life. Additionally, they can serve as a foundation for future studies on SCD in Iran.
Methods
Design and participants
This cross-sectional descriptive study was performed on individuals diagnosed with SCD who sought specialized and subspecialized care at clinics and were hospitalized in the wards of Bandar Abbas Children’s Hospital during the years 2022 to 2023.
Eligibility and ineligibility criteria
Inclusion criteria
• Age below 18 years
• Referral to specialized and sub-specialized heart and blood clinics or admission to the Children’s Hospital departments in Bandar Abbas City
• History of at least one episode of vascular occlusion crisis resulting in hospitalization
• Willingness to participate in the study
Exclusion criteria
• Non-native residents of the area
• Absence of prior hospitalization due to blood vessel obstruction
Sample selection
In this study, a census sampling method was used. Following the application of the inclusion and exclusion criteria, all eligible participants were enrolled.
Data collection
After obtaining informed consent and ethical clearance, eligible patients were assessed through a standardized checklist developed by the researcher. The checklist was validated by hematology and pediatric cardiology experts and included demographic data (age, gender) and the following clinical variables:
Pulmonary hypertension (PHT), hemoglobin levels, vascular occlusion crisis, high hematocrit levels, α-thalassemia , hydroxyurea treatment, white blood cell count reduction, high creatinine levels, genotype and high ferritin levels.
Data collection process
This checklist comprises crucial demographic information, encompassing gender and age, as well as pertinent treatment details, such as PHT, hemoglobin levels, vascular occlusion crisis, high hematocrit levels, α-thalassemia, hydroxyurea treatment, reduction of white blood cells, high creatinine levels, genotype, and high ferritin levels in affected individuals.
All participants underwent echocardiographic evaluation for PHT. Among individuals with sickle cell anemia, chronic anemia and the frequent development of cardiac complications contribute to PHT, which is considered one of the most severe complications associated with the disease. Relevant laboratory tests were conducted, and data were collected through interviews and a review of patient records.
Statistical analysis
Data were analyzed using SPSS software, version 22. Descriptive statistical methods, including Mean±SD, frequency, and percentage, were applied to describe the data. To assess the association between risk factors and the presence or absence of PHT, the chi-square test was employed.
Results
The research findings indicated that of the 75 patients assessed, 35 patients (46.7%) were female and 40 patients (53.3%) were male. The youngest age among the patients was 6 years, while the oldest age was 17 years. The mean age of the patients was determined to be 10.50±2.94 years.
Table 1 reveals that out of the individuals examined, 58 cases (77.3%) were determined to be free of PHT, whereas 17 cases (22.7%) were diagnosed with the condition.
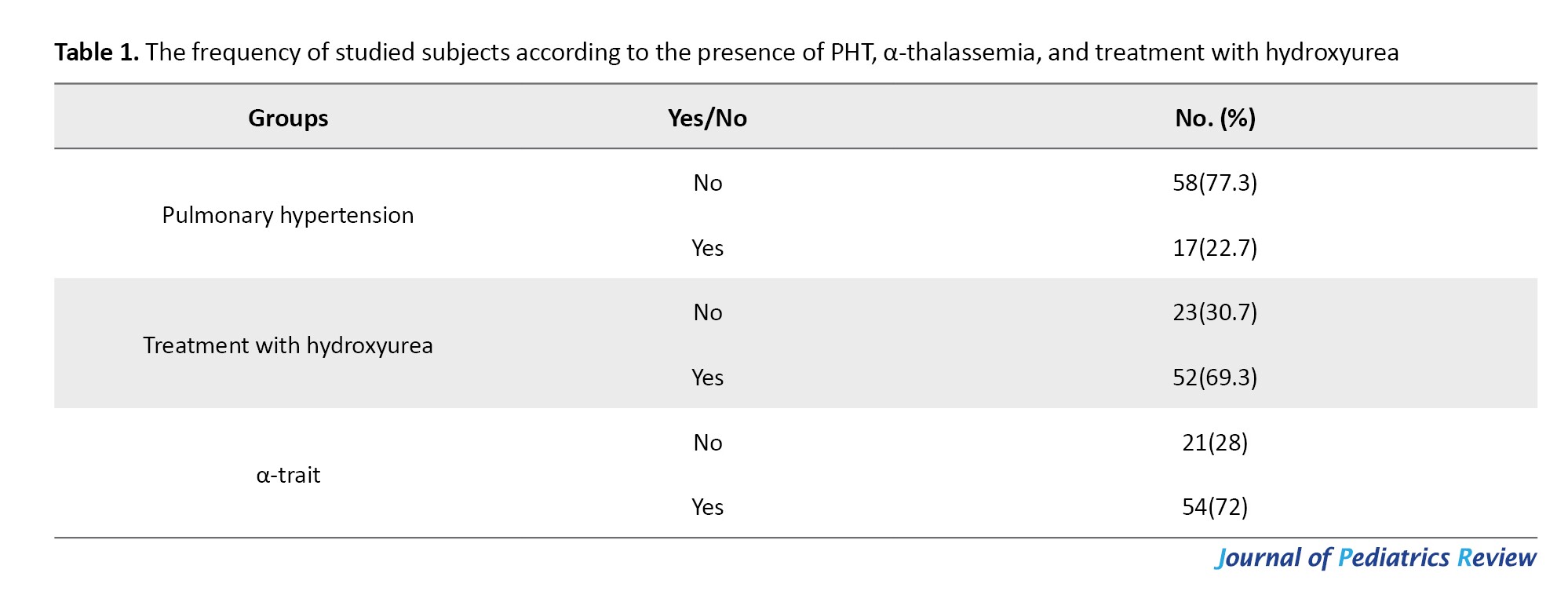
The findings indicate that 23 individuals (30.7%) did not receive hydroxyurea treatment, whereas the majority, 52 individuals (69.3%), were treated with hydroxyurea. Furthermore, a significant proportion of patients (72%) had α-thalassemia.
Table 2 reveals a comparison of gender based on the existence of PHT. Among those without PHT, 29 were female and 29 were male. On the other hand, among those with PHT, there were 11 females and 6 males. There was no statistically significant disparity between males and females in both the control and treatment groups `significant correlation was found between genotype and PHT (P>0.05).
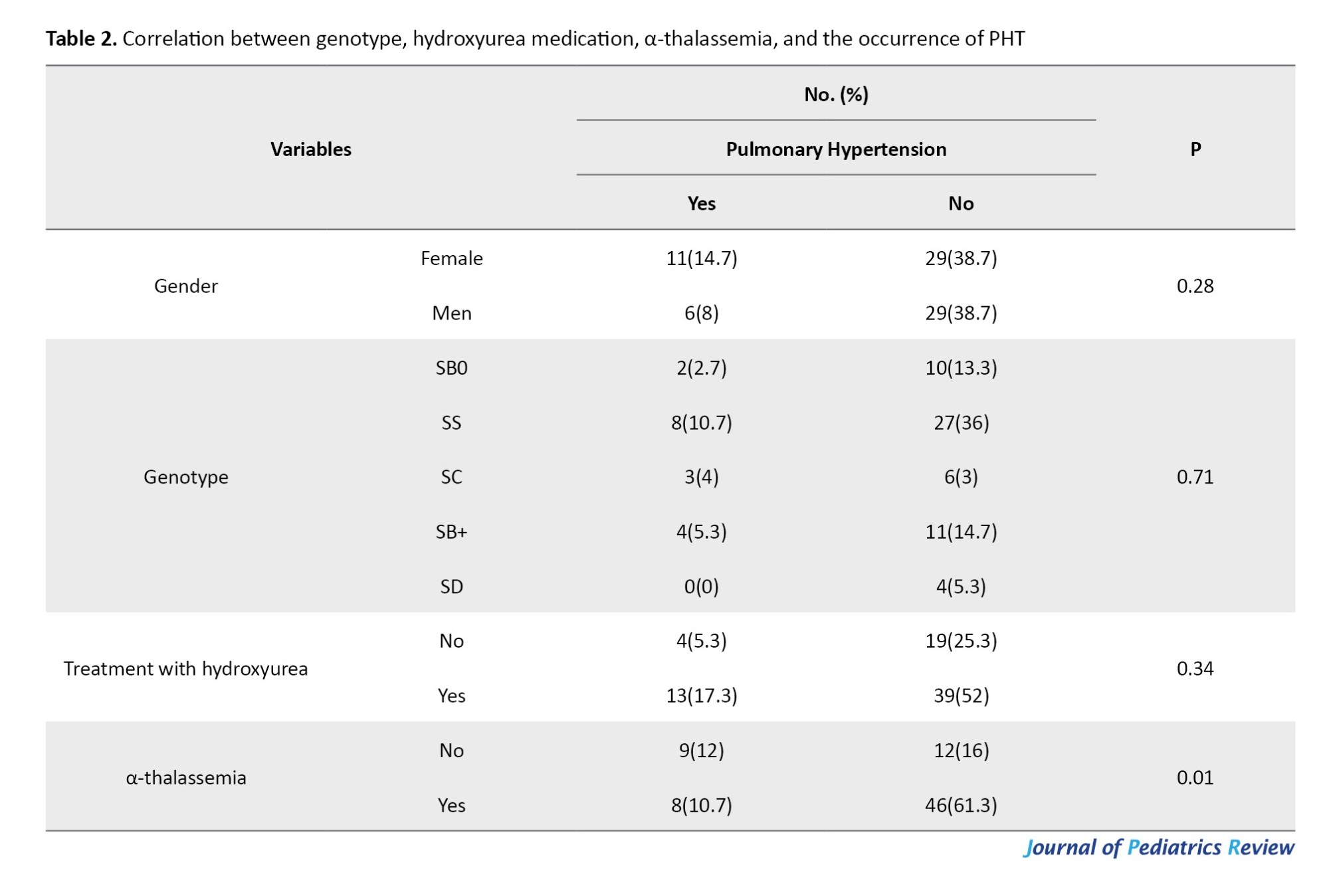
Based on the findings shown in Table 2, the majority of individuals (39 people) had used hydroxyurea but did not exhibit PHT. Conversely, a minority of individuals (4 people) had PHT but did not use hydroxyurea. There was no significant link between the usage of hydroxyurea and the occurrence of PHT (P>0.05). Additionally, a majority of individuals, specifically 12 individuals, did not exhibit α-thalassemia and PHT. Conversely, a minority of individuals, namely 8 individuals, exhibited α-thalassemia and PHT. There was a significant difference between α-thalassemia and the occurrence of PHT (P=0.01).
Table 3 presents a comparison of the correlation between age, hematocrit, platelets, WBC, hemoglobin levels, creatinine levels, ferritin levels, and the occurrence of PHT. Individuals with PHT had a greater mean age compared to the healthy individuals. However, this difference was not significant. The analysis of hematocrit data revealed that individuals with PHT had elevated hematocrit levels compared to those without disease, but this disparity was not significant (P>0.05).
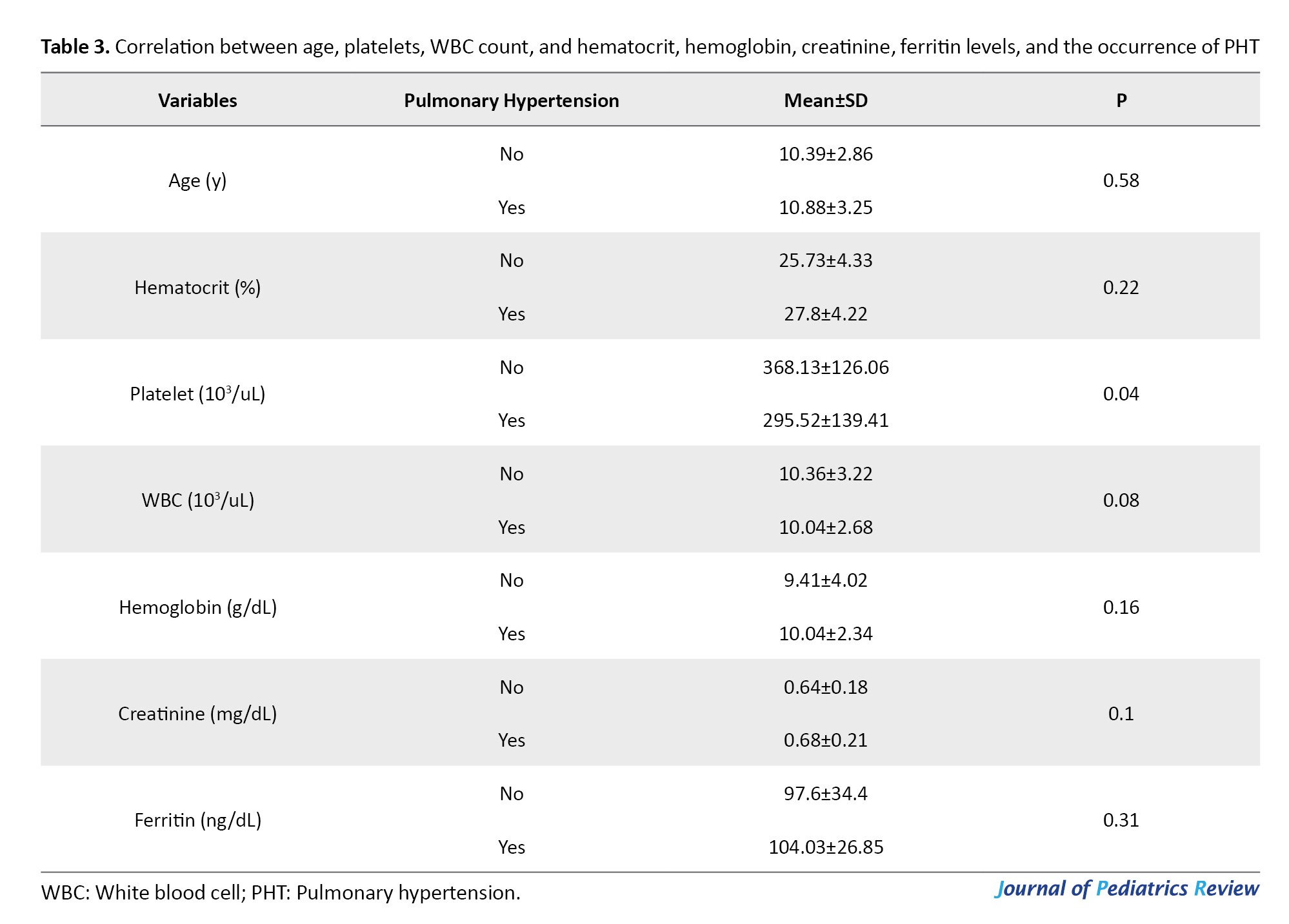
A statistically significant difference (P<0.05) was observed in platelet counts between patients with PHT and those without. Specifically, those who had the disease exhibited lower platelet counts. Additionally, individuals with PHT had a reduced count of leukocytes compared to those without the disease, but this difference was not significant (P>0.05). The analysis of haemoglobin, creatinine, and ferritin data revealed that individuals with PHT had elevated levels of hemoglobin, creatinine, and ferritin compared to healthy individuals. However, the increase was not significant (P>0.05).
Discussion
PHT is a serious and progressive complication in 6–10% of adults with SCD. While traditional mechanisms, like hemolysis, nitric oxide deficiency, and thrombosis, have been studied, the disease’s clinical variability remains unclear [9]. Due to its rapid global transmission, the World Health Organization (WHO) declared SARS-CoV-2, the virus responsible for acute respiratory syndrome, a pandemic in March 2020. Individuals with SCD have a heightened risk of experiencing severe clinical problems if they are infected with COVID-19 due to the widespread damage to their blood vessels and endothelial cells [10, 11]. Given this potential danger, it is crucial to methodically assess individuals with PHT in order to detect the many clinical presentations and consequences observed in children with SCD.
The findings of our study indicated that PHT was present in 22.7% of the patients diagnosed with sickle cell anemia. The study by Amadi et al. reported an overall prevalence of Doppler-derived PH of 38% among individuals with sickle cell anemia [12]. According to the research conducted by Elfaki et al., PHT was observed in 29% of the studied population [2]. Ramsey et al. found that PHT was prevalent among patients diagnosed with sickle cell anemia, a finding that aligns with the results of our study [13]. Sokunbi et al. revealed that the occurrence of PHT in children diagnosed with sickle cell anemia was found to be 22.9%, which aligns with the findings of our study [14]. Dosunmu et al., in a study conducted in Lagos, Nigeria, reported a significantly lower prevalence of 3.6%. This lower rate may be attributed to the age group of the study population compared to that of the present study [15].
In this cross-sectional study involving 75 Children affected by SCD, no significant correlation was found between age and gender and the occurrence of PHT in patients with SCD (P>0.05). Eskandarian et al. found no significant differences in age or gender between patients with and without PHT [16]. Tarrass et al. conducted an evaluation of PAP through echocardiography in patients undergoing dialysis. Their findings indicated no correlation between PHT and factors, such as age, gender, duration of dialysis, vascular access location, biological parameters, or parathyroid hormone levels [17].
Our study revealed that female patients and those of older age had a higher likelihood of developing PHT. According to the findings of Mukhtar et al., PHT was reported to occur more commonly in women; however, there was no significant relationship between gender and PHT [18]. Furthermore, our findings align with those of Odeyemi et al.’s study on children diagnosed with SCD, indicating that being female and advancing in age contribute to a higher occurrence of PHT in these individuals. In contrast to our investigation, this study found a statistically significant association (P<0.05) between gender, age, and the occurrence of PHT [19].
In our study, the predominant genotype among most participants was HbSS (SCD), accounting for 46.7% of the group. Elfaki et al. found a similar result, with 90% of their subjects carrying the HbSS genotype [2]. Our study identified a notable correlation between the α-thalassemia trait and the prevalence of PHT observed in SCD populations (P<0.05), indicating that those with α-thalassemia are less likely to develop PHT. Similarly, Fonseca et al. utilized echocardiography to assess pulmonary complications by evaluating right ventricular function, and their findings also demonstrated a significant association between α-thalassemia and a lower incidence of PHT (P<0.05) [20].
Fraidenburg and Machado reported that PHT is a very rare complication in individuals with α-thalassemia [21]. Furthermore, in another study that assessed pulmonary hemodynamics using echocardiography in patients with α-thalassemia, only 80.3(4%) of those diagnosed with HbH or Bart’s disease exhibited echocardiographic evidence of PHT, defined by a tricuspid regurgitant velocity exceeding 2.9 m/s [22]. Teawtrakul et al. indicated that patients with beta thalassemia had a significantly higher risk of developing PHT compared to those with α-thalassemia or combined α and β thalassemia (OR=9.47, P=0.036) [22]. The studies conducted by Dosunmu et al. and Aliyu et al. reported a significantly lower incidence of PHT in individuals with SCD who also possess the α-thalassemia (P<0.05), which is consistent with the findings of our research [4, 15].
The research findings demonstrated a significant correlation between PHTN and platelet counts (P<0.05), indicating that individuals with lower platelet counts had a higher likelihood of developing PHT. This conclusion is consistent with the findings of Sokunbi et al. [14]. The results of Liao and Wu indicated that red blood cell distribution width (RDW) and mean platelet volume (MPV) were significantly higher in patients with interstitial lung disease (ILD) and moderate-to-severe PHT (Ms-PH) compared to those with ILD without PHT and those with mild PHT (P<0.05). Furthermore, in patients with ILD, RDW, age, MPV, and serum IgG levels independently contributed to the risk of developing Ms-PH [23].
Hydroxyurea, which increases the generation of fetal hemoglobin (HbF), is a commonly used and successful therapy for SCD patients worldwide. An elevation in HbF expression helps reduce RBC sickling and decreases the vaso-occlusive crisis frequency [24, 25]. Consequently, the occurrence of vasculopathic consequences, such as PHT, is reduced, leading to a substantial increase in lifespan after a six-year course of therapy [26].
The findings revealed that the majority of individuals (39 people) had used hydroxyurea but did not show signs of PHT. In contrast, a smaller group (4 people) had PHT despite not using hydroxyurea. The data analysis revealed no significant correlation between the use of hydroxyurea and the existence of PHT. Elfaki et al. reported that, due to the high proportion of asymptomatic patients, initiating targeted treatment with hydroxyurea was not deemed warranted [2]. Consistent with our research, Sokunbi et al. and Klings et al. observed no significant correlation between hydroxyurea and PHT in their studies of individuals with SCD (P<0.05) [14, 25]. The lack of a significant correlation between PHTN and hydroxyurea administration in our research may be attributed to the delayed commencement or relatively short duration of this hydroxyurea therapy among our subjects. PHT was observed in a minority (17%) of the individuals who received hydroxyurea treatment.
The findings of the current study indicated no significant difference in mean hemoglobin levels between patients with and without PHT. Similarly, Sedighi et al., employing a 35 mm Hg threshold to diagnose PHT, reported no association between hemoglobin concentration and the occurrence of PHT [27]. In their study of pulmonary arterial pressure (PAP) in dialysis patients, Domenici et al. found that PHT was associated with longer dialysis durations, higher hemoglobin levels, and higher serum albumin levels compared to those with normal PAP [28]. Hemoglobin levels and the prevalence of PHT were shown to be highly associated in the study by Eskandarian et al. People with normal hemoglobin levels (≥11 g/dL) were approximately 70.2% less likely to develop PHT than those with low hemoglobin levels, according to the binary logistic regression analysis [16].
Conclusion
The study results showed that 22.7% of patients with SCD had PHT. PHT was significantly associated with α-thalassemia and platelet count. Additionally, the prevalence of the disease was higher in women than in men. Therefore, necessary diagnostic and preventive measures should be taken in high-risk individuals, considering the severe complications of the disease and the increased risk of mortality in these patients. It is advisable to conduct frequent screening for PHT in individuals with SCD, considering all pertinent criteria during the screening process.
Limitations of this research include the relatively small number of participants and its single-center design. Also, this study did not examine symptoms, such as smoking or current symptoms of the disease. It is suggested that future studies should examine more demographic and laboratory variables that may be influential.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Hormozgan University of Medical Sciences, Bandar abbas, Iran (Code: IR.HUMS.REC.1400.258). All participants or their legal guardians gave written informed consent prior to enrollment. Confidentiality and anonymity were strictly maintained by assigning participants unique identification codes and securely storing data. Also, participation was voluntary, and patients had the right to withdraw at any time without any repercussions for their medical care. The study also complied with the national ethical charter for biomedical research and the institutional ethical guidelines.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Authors contributions
All authors contributed equally to the conception and design of the study, data collection and analysis, interception of the results and drafting of the manuscript. Each author approved the final version of the manuscript for submission.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors express their heartfelt appreciation to the university research assistant, the hospital team, and the participants for their valuable assistance throughout this project.
References
Sickle cell disease (SCD) is an inherited disorder caused by an aberrant type of hemoglobin S (HbS), resulting from a single-point mutation in the β-globin gene [1, 2]. This condition is notably common in areas, including India, Sub-Saharan Africa, Saudi Arabia, and South America. Individuals affected by SCD inherit two copies of the HbS gene, leading to hemolytic anemia and complications such as acute chest syndrome and vaso-occlusive pain crises (VOC). Because HbS precipitates during deoxygenation, symptoms usually manifest within the first year of life, causing erythrocytes to assume a sickle shape. This deformation disrupts blood flow, leading to hemolysis and vaso-occlusive episodes [1-3]. SCD patients are susceptible to progressive vasculopathy, which is characterized by endothelial dysfunction, systemic issues, PHT, and structural alterations, such as smooth muscle remodeling and intimal proliferation in blood vessels [1].
PHT represents a serious and potentially fatal complication of SCD, impacting the pulmonary vessels and compromising the structure and function of the right ventricle. It is characterized by a resting mean pulmonary artery pressure of more than 25 mm Hg. Notwithstanding its clinical significance, there is a lack of clarity regarding the prevalence and risk factors of PHT in individuals with SCD, and differences have been noted between studies and groups [4, 5].
PHT in SCD is associated with a range of crippling consequences, such as a worse quality of life, decreased ability to exercise, trouble with day-to-day functioning, and noticeably lower survival rates [6]. Common clinical symptoms include fatigue, peripheral edema, dyspnea, and chest pain. During the physical examination, one might observe indicators of right ventricular dysfunction, including a pronounced second heart sound, evidence of right ventricular hypertrophy, right-sided fourth heart sound, and right parasternal heave. The findings provide valuable understanding into cardiovascular functional and structural changes, aiding in diagnosis and guiding targeted management strategies [2].
The diagnosis of PHT in SCD patients typically depends on invasive right cardiac catheterization, regarded as the gold standard for assessing pulmonary artery pressure. Noninvasive methods, especially Doppler echocardiography, are employed to evaluate right ventricular dimensions and performance while estimating pulmonary artery systolic pressure (PASP). As a valuable screening tool, Doppler echocardiography helps identify people susceptible to PHT, enabling additional assessment and action [7]. Acute complications of SCD, such as vaso-occlusive crises and acute chest syndrome, can exacerbate PHT, resulting in rapid clinical decline and potentially life-threatening consequences. Thus, early identification and prompt intervention in SCD patients with PHT are essential to enhance prognosis and optimize patient outcomes [8].
SCD is one of the most common hemoglobinopathies in Iran, particularly in the southern regions. It is associated with vascular occlusive crises, severe pain, and multisystem complications; thus, studying its complications is of significant importance. However, data on the prevalence, incidence, and associated factors of PHT in the Iranian population, especially among children and adolescents under 18 years of age, are limited. This study, utilizing echocardiography and analyzing data from hospitalized patients, aimed to determine the incidence of this complication and examine its association with demographic and treatment-related factors. Therefore, the study was conducted to investigate the incidence of PHT in patients with SCD in Bandar Abbas in 2022-2023. The findings of this research could play a crucial role in early diagnosis, effective therapeutic interventions, and the development of management strategies to improve patients’ quality of life. Additionally, they can serve as a foundation for future studies on SCD in Iran.
Methods
Design and participants
This cross-sectional descriptive study was performed on individuals diagnosed with SCD who sought specialized and subspecialized care at clinics and were hospitalized in the wards of Bandar Abbas Children’s Hospital during the years 2022 to 2023.
Eligibility and ineligibility criteria
Inclusion criteria
• Age below 18 years
• Referral to specialized and sub-specialized heart and blood clinics or admission to the Children’s Hospital departments in Bandar Abbas City
• History of at least one episode of vascular occlusion crisis resulting in hospitalization
• Willingness to participate in the study
Exclusion criteria
• Non-native residents of the area
• Absence of prior hospitalization due to blood vessel obstruction
Sample selection
In this study, a census sampling method was used. Following the application of the inclusion and exclusion criteria, all eligible participants were enrolled.
Data collection
After obtaining informed consent and ethical clearance, eligible patients were assessed through a standardized checklist developed by the researcher. The checklist was validated by hematology and pediatric cardiology experts and included demographic data (age, gender) and the following clinical variables:
Pulmonary hypertension (PHT), hemoglobin levels, vascular occlusion crisis, high hematocrit levels, α-thalassemia , hydroxyurea treatment, white blood cell count reduction, high creatinine levels, genotype and high ferritin levels.
Data collection process
This checklist comprises crucial demographic information, encompassing gender and age, as well as pertinent treatment details, such as PHT, hemoglobin levels, vascular occlusion crisis, high hematocrit levels, α-thalassemia, hydroxyurea treatment, reduction of white blood cells, high creatinine levels, genotype, and high ferritin levels in affected individuals.
All participants underwent echocardiographic evaluation for PHT. Among individuals with sickle cell anemia, chronic anemia and the frequent development of cardiac complications contribute to PHT, which is considered one of the most severe complications associated with the disease. Relevant laboratory tests were conducted, and data were collected through interviews and a review of patient records.
Statistical analysis
Data were analyzed using SPSS software, version 22. Descriptive statistical methods, including Mean±SD, frequency, and percentage, were applied to describe the data. To assess the association between risk factors and the presence or absence of PHT, the chi-square test was employed.
Results
The research findings indicated that of the 75 patients assessed, 35 patients (46.7%) were female and 40 patients (53.3%) were male. The youngest age among the patients was 6 years, while the oldest age was 17 years. The mean age of the patients was determined to be 10.50±2.94 years.
Table 1 reveals that out of the individuals examined, 58 cases (77.3%) were determined to be free of PHT, whereas 17 cases (22.7%) were diagnosed with the condition.
The findings indicate that 23 individuals (30.7%) did not receive hydroxyurea treatment, whereas the majority, 52 individuals (69.3%), were treated with hydroxyurea. Furthermore, a significant proportion of patients (72%) had α-thalassemia.
Table 2 reveals a comparison of gender based on the existence of PHT. Among those without PHT, 29 were female and 29 were male. On the other hand, among those with PHT, there were 11 females and 6 males. There was no statistically significant disparity between males and females in both the control and treatment groups `significant correlation was found between genotype and PHT (P>0.05).
Based on the findings shown in Table 2, the majority of individuals (39 people) had used hydroxyurea but did not exhibit PHT. Conversely, a minority of individuals (4 people) had PHT but did not use hydroxyurea. There was no significant link between the usage of hydroxyurea and the occurrence of PHT (P>0.05). Additionally, a majority of individuals, specifically 12 individuals, did not exhibit α-thalassemia and PHT. Conversely, a minority of individuals, namely 8 individuals, exhibited α-thalassemia and PHT. There was a significant difference between α-thalassemia and the occurrence of PHT (P=0.01).
Table 3 presents a comparison of the correlation between age, hematocrit, platelets, WBC, hemoglobin levels, creatinine levels, ferritin levels, and the occurrence of PHT. Individuals with PHT had a greater mean age compared to the healthy individuals. However, this difference was not significant. The analysis of hematocrit data revealed that individuals with PHT had elevated hematocrit levels compared to those without disease, but this disparity was not significant (P>0.05).
A statistically significant difference (P<0.05) was observed in platelet counts between patients with PHT and those without. Specifically, those who had the disease exhibited lower platelet counts. Additionally, individuals with PHT had a reduced count of leukocytes compared to those without the disease, but this difference was not significant (P>0.05). The analysis of haemoglobin, creatinine, and ferritin data revealed that individuals with PHT had elevated levels of hemoglobin, creatinine, and ferritin compared to healthy individuals. However, the increase was not significant (P>0.05).
Discussion
PHT is a serious and progressive complication in 6–10% of adults with SCD. While traditional mechanisms, like hemolysis, nitric oxide deficiency, and thrombosis, have been studied, the disease’s clinical variability remains unclear [9]. Due to its rapid global transmission, the World Health Organization (WHO) declared SARS-CoV-2, the virus responsible for acute respiratory syndrome, a pandemic in March 2020. Individuals with SCD have a heightened risk of experiencing severe clinical problems if they are infected with COVID-19 due to the widespread damage to their blood vessels and endothelial cells [10, 11]. Given this potential danger, it is crucial to methodically assess individuals with PHT in order to detect the many clinical presentations and consequences observed in children with SCD.
The findings of our study indicated that PHT was present in 22.7% of the patients diagnosed with sickle cell anemia. The study by Amadi et al. reported an overall prevalence of Doppler-derived PH of 38% among individuals with sickle cell anemia [12]. According to the research conducted by Elfaki et al., PHT was observed in 29% of the studied population [2]. Ramsey et al. found that PHT was prevalent among patients diagnosed with sickle cell anemia, a finding that aligns with the results of our study [13]. Sokunbi et al. revealed that the occurrence of PHT in children diagnosed with sickle cell anemia was found to be 22.9%, which aligns with the findings of our study [14]. Dosunmu et al., in a study conducted in Lagos, Nigeria, reported a significantly lower prevalence of 3.6%. This lower rate may be attributed to the age group of the study population compared to that of the present study [15].
In this cross-sectional study involving 75 Children affected by SCD, no significant correlation was found between age and gender and the occurrence of PHT in patients with SCD (P>0.05). Eskandarian et al. found no significant differences in age or gender between patients with and without PHT [16]. Tarrass et al. conducted an evaluation of PAP through echocardiography in patients undergoing dialysis. Their findings indicated no correlation between PHT and factors, such as age, gender, duration of dialysis, vascular access location, biological parameters, or parathyroid hormone levels [17].
Our study revealed that female patients and those of older age had a higher likelihood of developing PHT. According to the findings of Mukhtar et al., PHT was reported to occur more commonly in women; however, there was no significant relationship between gender and PHT [18]. Furthermore, our findings align with those of Odeyemi et al.’s study on children diagnosed with SCD, indicating that being female and advancing in age contribute to a higher occurrence of PHT in these individuals. In contrast to our investigation, this study found a statistically significant association (P<0.05) between gender, age, and the occurrence of PHT [19].
In our study, the predominant genotype among most participants was HbSS (SCD), accounting for 46.7% of the group. Elfaki et al. found a similar result, with 90% of their subjects carrying the HbSS genotype [2]. Our study identified a notable correlation between the α-thalassemia trait and the prevalence of PHT observed in SCD populations (P<0.05), indicating that those with α-thalassemia are less likely to develop PHT. Similarly, Fonseca et al. utilized echocardiography to assess pulmonary complications by evaluating right ventricular function, and their findings also demonstrated a significant association between α-thalassemia and a lower incidence of PHT (P<0.05) [20].
Fraidenburg and Machado reported that PHT is a very rare complication in individuals with α-thalassemia [21]. Furthermore, in another study that assessed pulmonary hemodynamics using echocardiography in patients with α-thalassemia, only 80.3(4%) of those diagnosed with HbH or Bart’s disease exhibited echocardiographic evidence of PHT, defined by a tricuspid regurgitant velocity exceeding 2.9 m/s [22]. Teawtrakul et al. indicated that patients with beta thalassemia had a significantly higher risk of developing PHT compared to those with α-thalassemia or combined α and β thalassemia (OR=9.47, P=0.036) [22]. The studies conducted by Dosunmu et al. and Aliyu et al. reported a significantly lower incidence of PHT in individuals with SCD who also possess the α-thalassemia (P<0.05), which is consistent with the findings of our research [4, 15].
The research findings demonstrated a significant correlation between PHTN and platelet counts (P<0.05), indicating that individuals with lower platelet counts had a higher likelihood of developing PHT. This conclusion is consistent with the findings of Sokunbi et al. [14]. The results of Liao and Wu indicated that red blood cell distribution width (RDW) and mean platelet volume (MPV) were significantly higher in patients with interstitial lung disease (ILD) and moderate-to-severe PHT (Ms-PH) compared to those with ILD without PHT and those with mild PHT (P<0.05). Furthermore, in patients with ILD, RDW, age, MPV, and serum IgG levels independently contributed to the risk of developing Ms-PH [23].
Hydroxyurea, which increases the generation of fetal hemoglobin (HbF), is a commonly used and successful therapy for SCD patients worldwide. An elevation in HbF expression helps reduce RBC sickling and decreases the vaso-occlusive crisis frequency [24, 25]. Consequently, the occurrence of vasculopathic consequences, such as PHT, is reduced, leading to a substantial increase in lifespan after a six-year course of therapy [26].
The findings revealed that the majority of individuals (39 people) had used hydroxyurea but did not show signs of PHT. In contrast, a smaller group (4 people) had PHT despite not using hydroxyurea. The data analysis revealed no significant correlation between the use of hydroxyurea and the existence of PHT. Elfaki et al. reported that, due to the high proportion of asymptomatic patients, initiating targeted treatment with hydroxyurea was not deemed warranted [2]. Consistent with our research, Sokunbi et al. and Klings et al. observed no significant correlation between hydroxyurea and PHT in their studies of individuals with SCD (P<0.05) [14, 25]. The lack of a significant correlation between PHTN and hydroxyurea administration in our research may be attributed to the delayed commencement or relatively short duration of this hydroxyurea therapy among our subjects. PHT was observed in a minority (17%) of the individuals who received hydroxyurea treatment.
The findings of the current study indicated no significant difference in mean hemoglobin levels between patients with and without PHT. Similarly, Sedighi et al., employing a 35 mm Hg threshold to diagnose PHT, reported no association between hemoglobin concentration and the occurrence of PHT [27]. In their study of pulmonary arterial pressure (PAP) in dialysis patients, Domenici et al. found that PHT was associated with longer dialysis durations, higher hemoglobin levels, and higher serum albumin levels compared to those with normal PAP [28]. Hemoglobin levels and the prevalence of PHT were shown to be highly associated in the study by Eskandarian et al. People with normal hemoglobin levels (≥11 g/dL) were approximately 70.2% less likely to develop PHT than those with low hemoglobin levels, according to the binary logistic regression analysis [16].
Conclusion
The study results showed that 22.7% of patients with SCD had PHT. PHT was significantly associated with α-thalassemia and platelet count. Additionally, the prevalence of the disease was higher in women than in men. Therefore, necessary diagnostic and preventive measures should be taken in high-risk individuals, considering the severe complications of the disease and the increased risk of mortality in these patients. It is advisable to conduct frequent screening for PHT in individuals with SCD, considering all pertinent criteria during the screening process.
Limitations of this research include the relatively small number of participants and its single-center design. Also, this study did not examine symptoms, such as smoking or current symptoms of the disease. It is suggested that future studies should examine more demographic and laboratory variables that may be influential.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Hormozgan University of Medical Sciences, Bandar abbas, Iran (Code: IR.HUMS.REC.1400.258). All participants or their legal guardians gave written informed consent prior to enrollment. Confidentiality and anonymity were strictly maintained by assigning participants unique identification codes and securely storing data. Also, participation was voluntary, and patients had the right to withdraw at any time without any repercussions for their medical care. The study also complied with the national ethical charter for biomedical research and the institutional ethical guidelines.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Authors contributions
All authors contributed equally to the conception and design of the study, data collection and analysis, interception of the results and drafting of the manuscript. Each author approved the final version of the manuscript for submission.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors express their heartfelt appreciation to the university research assistant, the hospital team, and the participants for their valuable assistance throughout this project.
References
- Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: A review. JAMA. 2022; 328(1):57-68. [DOI:10.1001/jama.2022.10233] [PMID]
- Elfaki YA, Ibrahim AS, Merghani TH. Prevalence of pulmonary hypertension among sudanese patients with sickle cell disease. Open Respir Med J. 2024; 18:e18743064292252. [DOI:10.2174/0118743064292252240422100911] [PMID] [PMCID]
- Payne AB, Mehal JM, Chapman C, Haberling DL, Richardson LC, Bean CJ, et al. Trends in sickle cell disease-related mortality in the United States, 1979 to 2017. Ann Emerg Med. 2020; 76(3S):S28-36. [DOI:10.1016/j.annemergmed.2020.08.009] [PMID]
- Aliyu ZY, Gordeuk V, Sachdev V, Babadoko A, Mamman AI, Akpanpe P, et al. Prevalence and risk factors for pulmonary artery systolic hypertension among sickle cell disease patients in Nigeria. Am J Hematol. 2008; 83(6):485-90. [DOI:10.1002/ajh.21162] [PMID] [PMCID]
- Ondze Kafata LI NL, Letomo KN, et al. Echocardiographic aspects of Congolese sickle cell disease heart. Arch Cardiovasc Dis Suppl. 2016; 8(1):43. [DOI:10.1016/S1878-6480(16)30132-X]
- Hammoudi N, Lionnet F, Redheuil A, Montalescot G. Cardiovascular manifestations of sickle cell disease. Eur Heart J. 2020; 41(13):1365-73. [DOI:10.1093/eurheartj/ehz217] [PMID]
- Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, et al. Guidelines for the echocardiographic assessment of the right heart in adults: A report from the American society of echocardiography: Endorsed by the European association of echocardiography, a registered branch of the European society of cardiology, and the Canadian society of echocardiography. J Am Soc Echocardiogr. 2010; 23(7):685-713. [DOI:10.1016/j.echo.2010.05.010] [PMID]
- Yousef AA, Shash HA, Almajid AN, Binammar AA, Almusabeh HA, Alshaqaq HM,et al. Acute chest syndrome in pediatric sickle cell disease: A 19-year tertiary center experience. Ann Thorac Med. 2022; 17(4):199-206. [DOI:10.4103/atm.atm_575_21] [PMID] [PMCID]
- Chatzidavid S, Flevari P, Tombrou I, Anastasiadis G, Dimopoulou M; International hemoglobinopathy research network (INHERENT). Pulmonary hypertension in sickle cell disease: Novel findings of gene polymorphisms related to pathophysiology. Int J Mol Sci. 2024; 25(9):4792. [DOI:10.3390/ijms25094792] [PMID] [PMCID]
- Deldar M, Ghorabi ST, Sayehmiri K. SIR model for estimations of the coronavirus epidemic dynamics in Iran. J Biostat Epidemiol. 2020; 6(2):101-6. [Link]
- Hussain FA, Njoku FU, Saraf SL, Molokie RE, Gordeuk VR, Han J. COVID-19 infection in patients with sickle cell disease. Br J Haematol. 2020; 189(5):851-2. [DOI:10.1111/bjh.16734] [PMID] [PMCID]
- Amadi VN, Balogun MO, Akinola NO, Adebayo RA, Akintomide AO. Pulmonary hypertension in Nigerian adults with sickle cell anemia. Vasc Health Risk Manag. 2017; 13:153-60. [DOI:10.2147/VHRM.S92799] [PMID] [PMCID]
- Ramsey SD, Bender MA, Li L, Johnson KM, Jiao B, Devine B, et al. Prevalence of comorbidities associated with sickle cell disease among non-elderly individuals with commercial insurance-A retrospective cohort study. Plos One. 2022; 17(11):e0278137. [DOI:10.1371/journal.pone.0278137] [PMID] [PMCID]
- Sokunbi OJ, Ekure EN, Temiye EO, Anyanwu R, Okoromah CAN. Pulmonary hypertension among 5 to 18 year old children with sickle cell anaemia in Nigeria. Plos One. 2017; 12(9):e0184287. [DOI:10.1371/journal.pone.0184287] [PMID] [PMCID]
- Dosunmu AO, Balogun TM, Adeyeye OO, Daniel FA, Akinola RA, Onakoya JA, et al. Prevalence of pulmonary hypertension in sickle cell anaemia patients of a tertiary hospital in Nigeria. Niger Med J. 2014; 55(2):161-5. [DOI:10.4103/0300-1652.129661] [PMID] [PMCID]
- Eskandarian R SM, Yarmohamadi M, Mirmohammadkhani M. Pulmonary artery pressure status in hemodialysis patients and its association with nutritional and biochemicalmarkers. Middle East J Rehabil Health Stud. 2022; 11(3):e129698. [DOI:10.5812/mejrh-129698]
- Tarrass F, Benjelloun M, Medkouri G, Hachim K, Benghanem MG, Ramdani B. Doppler echocardiograph evaluation of pulmonary hypertension in patients undergoing hemodialysis. Hemodial Int. 2006; 10(4):356-9. [DOI:10.1111/j.1542-4758.2006.00129.x] [PMID]
- Mukhtar KN, Mohkumuddin S, Mahmood SN. Frequency of pulmonary hypertension in hemodialysis patients. Pak J Med Sci. 2014; 30(6):1319-22. [DOI:10.12669/pjms.306.5525] [PMID] [PMCID]
- Odeyemi AO, Oni OO, Odeyemi AO, Olufemi-Aworinde KJ, Ala OA, Abolarin AT. Pulmonary hypertension in people with sickle cell disease in a Nigerian tertiary hospital. Assam J Intern Med. 2022; 12(1):3-9. [DOI:10.4103/ajoim.ajoim_23_21]
- Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J. 2012; 39(1):112-8. [DOI:10.1183/09031936.00134410] [PMID]
- Fraidenburg DR, Machado RF. Pulmonary hypertension associated with thalassemia syndromes. Ann N Y Acad Sci. 2016; 1368(1):127-39. [DOI:10.1111/nyas.13037] [PMID] [PMCID]
- Teawtrakul N, Ungprasert P, Pussadhamma B, Prayalaw P, Fucharoen S, Jetsrisuparb A, et al. Effect of genotype on pulmonary hypertension risk in patients with thalassemia. Eur J Haematol. 2014; 92(5):429-34. [DOI:10.1111/ejh.12261] [PMID]
- Liao Y, Wu B. Analysis of clinical features and risk factors of pulmonary hypertension associated with interstitial lung disease. Biomed Rep. 2025; 22(4):58. [DOI:10.3892/br.2025.1936] [PMID] [PMCID]
- Sheikh AB, Nasrullah A, Lopez ED, Tanveer Ud Din M, Sagheer S, et al. Sickle cell disease-induced pulmonary hypertension: A review of pathophysiology, management, and current literature. Pulse. 2021; 9(3-4):57-63. [DOI:10.1159/000519101] [PMID] [PMCID]
- Klings ES, Machado RF, Barst RJ, Morris CR, Mubarak KK, Gordeuk VR, et al. An official American Thoracic Society clinical practice guideline: Diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med. 2014; 189(6):727-40. [DOI:10.1164/rccm.201401-0065ST] [PMID] [PMCID]
- Lobo CL, Pinto JF, Nascimento EM, Moura PG, Cardoso GP, Hankins JS. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br J Haematol. 2013; 161(6):852-60. [DOI:10.1111/bjh.12323] [PMID]
- Sedighi O, Golshani S, Sharifpoor A, Mahjoob F. The prevalence of pulmonary hypertension and the related factors in hemodialysis patients. J Mazandaran Univ Med Sci. 2011; 21(85):48-53. [Link]
- Domenici A, Luciani R, Principe F. Pulmonary hypertension in dialysis patients. Perit Dial Int. 2010; 30(2):251-2. [DOI:10.3747/pdi.2009.00082] [PMID]
Type of Study: Original Article |
Subject:
Pediatrics
Received: 2024/04/17 | Accepted: 2025/07/19 | Published: 2025/07/19
Received: 2024/04/17 | Accepted: 2025/07/19 | Published: 2025/07/19
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
