
Sun, Jun 14, 2026


Volume 12, Issue 4 (10-2024)
J. Pediatr. Rev 2024, 12(4): 359-368 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Zhang Y, Wang Y, Zhang J, Song N. Three Cases of Pigmentary Incontinence and Literature Review. J. Pediatr. Rev 2024; 12 (4) :359-368
URL: http://jpr.mazums.ac.ir/article-1-658-en.html
URL: http://jpr.mazums.ac.ir/article-1-658-en.html



1- Department of Neonatology, The Second Hospital of Jilin University, Changchun, China. , zhangyunf@jlu.edu.cn
2- Department of Neonatology, The Second Hospital of Jilin University, Changchun, China.
2- Department of Neonatology, The Second Hospital of Jilin University, Changchun, China.
Full-Text [PDF 1730 kb]
(835 Downloads)
| Abstract (HTML) (1748 Views)
Full-Text: (761 Views)
Introduction
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, with a prevalence of 0.7-1.2/100 000 [1, 2], is an X-chromosome-linked dominant disorder caused by mutations in the inhibitor of nuclear factor kappa-B kinase subunit gamma (NEMO/IKBKG) gene mutation at Xq28 [1]. More than 75% of IP patients are sporadic cases and the rest are familial about 10-25% [3]. It is usually fatal for male fetuses, mostly dying in utero [1]. In rare cases, male IP patients can survive if combined with Klinefelter syndrome (47, XXY karyotype), subtype mutations, or IKBKG somatic mosaicism [1, 2, 4, 5].
Case Presentation
Case number 1
A female patient was admitted to our hospital with a postnatal rash. The patient was delivered by cesarean section at a gestational age of 39 weeks with a birth weight of 3600 g. A family history of skin condition was denied. The mother had a 13-year-old daughter with no history of rash. After birth, the patient was found to have scattered pustules all over the skin, which protruded from the skin surface, with no obvious ulceration or exudation. Blood routine test results showed a leukocyte count of 40.8×109/L, a neutrophil percentage of 76.5%, a neutrophil count of 31.20×109/L, an eosinophil percentage of 7.3% and an eosinophil count of 1.40×109/L. The patient was administered cefepime for anti-infective treatment. On the second day of admission, most of the generalized rash subsided, leaving hyperpigmentation and dandruff and scattered herpes was still seen on the limbs, and the routine blood test results showed that the
white blood cell count was 20.6×109/L, the percentage of neutrophil was 58.2%, the neutrophil count was 12.01×109/L, the percentage of eosinophils was 2.04%, and the count of eosinophils was 0.10×109/L. On the fifth day of admission, red papules appeared on the limbs, and after consultation with the doctor from the Department of Dermatology, the diagnosis of IP was assigned. Systemic application of antibiotics was discontinued and topical skin healing film (2-3 times/day) was administered to prevent skin infections. Fundus examination and fundus fluorescein angiography (FFA) showed ischemic changes in the retina of both eyes (Figure 1 A-D), and retinal laser photocoagulation was performed in both eyes.
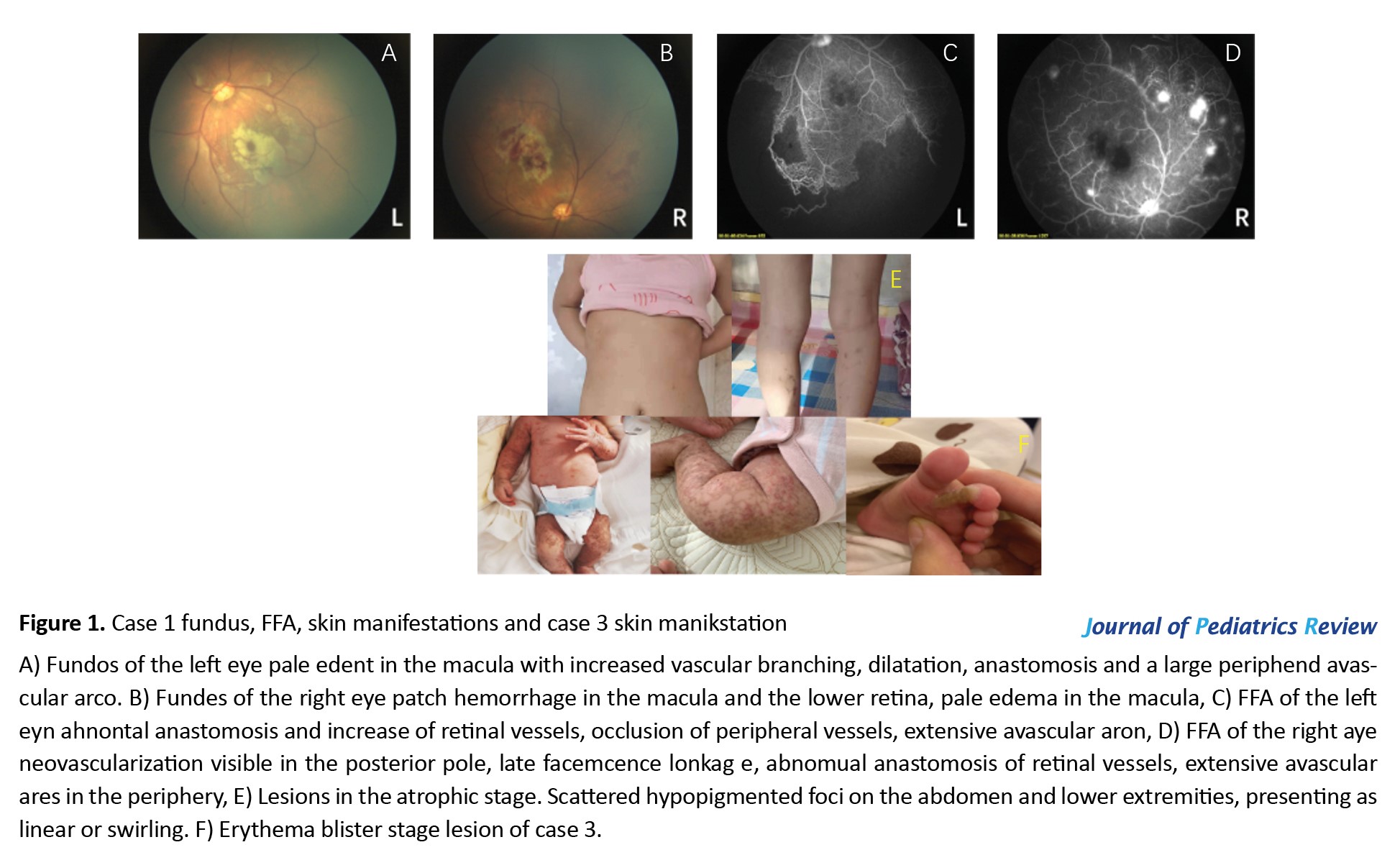 The genetic examination result was negative, and no abnormality was found in the brain and heart ultrasound as well as head nuclear magnetic resonance imaging (MRI). After the manuscript, the patient was 7 years old, with only scattered hypopigmentation foci on the abdomen and bilateral posterior calves (Figure 1E), accompanied by strabismus, missing teeth, and normal growth and development.
The genetic examination result was negative, and no abnormality was found in the brain and heart ultrasound as well as head nuclear magnetic resonance imaging (MRI). After the manuscript, the patient was 7 years old, with only scattered hypopigmentation foci on the abdomen and bilateral posterior calves (Figure 1E), accompanied by strabismus, missing teeth, and normal growth and development.
Case number 2
A 4-month-old female infant was diagnosed with IP on skin biopsy at another hospital (Figure 2A).
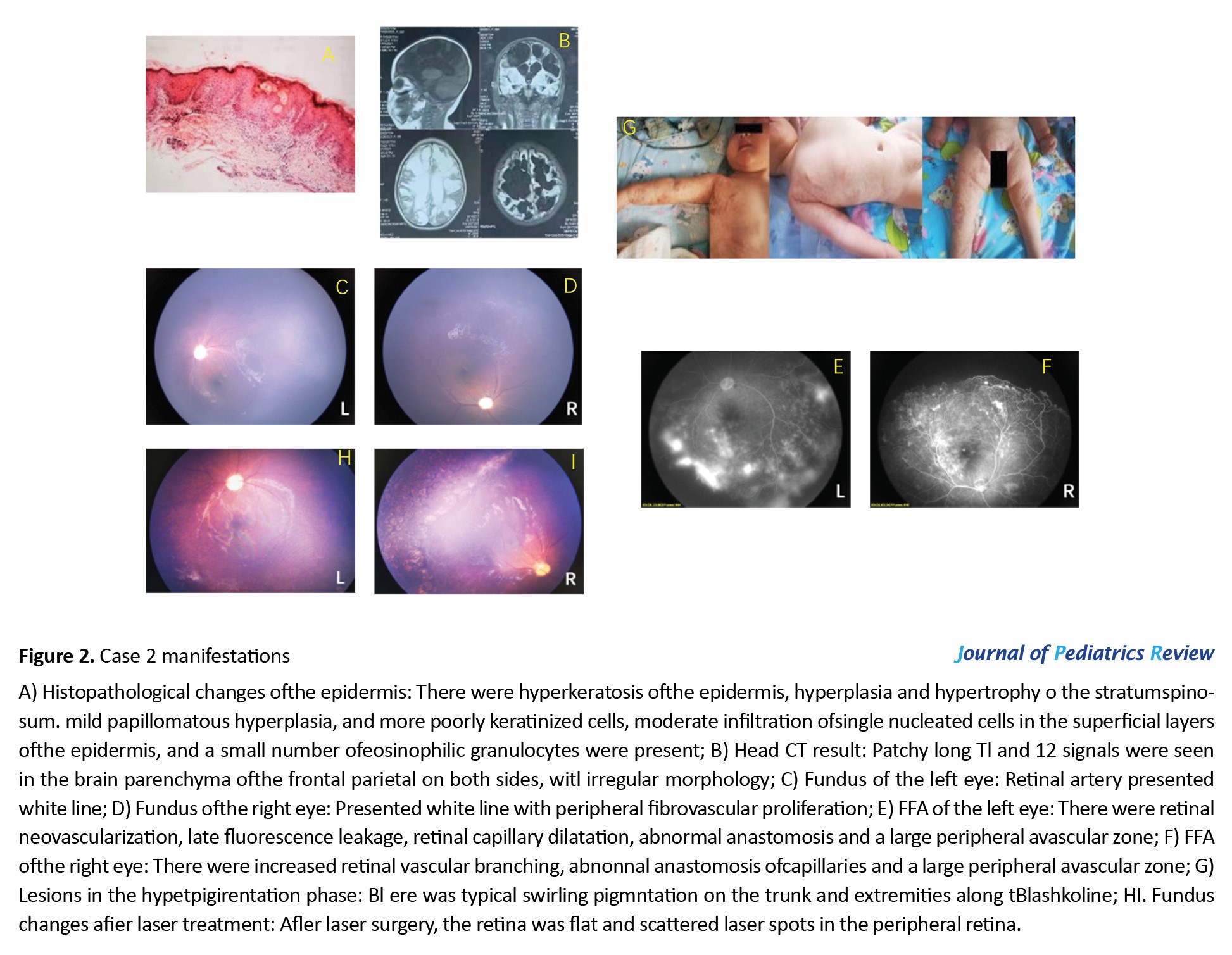 Head MRI showed large soft foci in the frontal parietal area bilaterally (Figure 2B). To check her fundus and FFA (Figure 2 C-F) and receive retinal laser photocoagulation, the patient presented to our department at the age of 8 months. The infant showed growth retardation, which was significantly improved after rehabilitation training. At a follow-up visit at the age of 11 months, typical swirling pigmentation was seen on the trunk and four limbs (Figure 2G). The fundus presented post-laser therapy changes (Figure 2 H-I).
Head MRI showed large soft foci in the frontal parietal area bilaterally (Figure 2B). To check her fundus and FFA (Figure 2 C-F) and receive retinal laser photocoagulation, the patient presented to our department at the age of 8 months. The infant showed growth retardation, which was significantly improved after rehabilitation training. At a follow-up visit at the age of 11 months, typical swirling pigmentation was seen on the trunk and four limbs (Figure 2G). The fundus presented post-laser therapy changes (Figure 2 H-I).
Case number 3
A female infant, presented to our department 20 days after birth with a rash (Figure 1F), whose mother was an IP patient with a history of miscarriage of male fetus. A complete head MRI of fundus cardiac color showed no abnormality. She is currently under regular follow-up.
Discussion
IP is an ectodermal disease that involves multiple systems and is characterized by skin pigmentation accompanied by damage to the teeth, eyes, central nervous system, mammary glands, and bones [1]. The following is an overview of the presentation and treatment of IP for each system.
IP skin performance
Skin changes are the most typical manifestations of IP, and the diagnosis can be confirmed based on skin manifestations alone [1]. The cutaneous manifestations occur sequentially in four stages, not all of which are mandatory and may overlap. The first stage is characterized by blisters distributed along Blaschko’s line, which most often occur in the neonatal period and may be accompanied by an elevated peripheral blood leukocyte count and eosinophil count. The rash may recur during febrile illnesses, infections (coxsackievirus) and vaccinations [6]. In the second stage, the blisters heal and form warty lesions and hyperkeratosis. The third stage is the hyperpigmentation phase, which can last until adulthood. The last stage mostly occurs before the complete disappearance of hyperpigmentation and is characterized by pale, hairless lesions [1].
Hadj-Rabia et al. [7] described the pathohistological changes in the skin at each stage and proposed them as secondary criteria for IP. Adults with IP may no longer have typical skin manifestations [8]. The manifestations of gray-blue mottled alopecia without capillary dilatation and follicular exfoliation/clogging found on dermoscopy can help in the diagnosis of IP. Skin damage usually resolves spontaneously without treatment, and in a few severe cases, it can be treated with topical hormonal therapy [3]. Both case 1 and case 2 reported in this paper had skin lesions combined with skin infections, which gradually healed after administration of systemic anti-infection therapy. In neonates, topical sterile dressings can be applied to prevent infections [3], and the skin healing film applied in case 1 is a sterile dressing, which achieved good results in case 1. Laser treatment can be attempted for recurrent inflammation, while avoidance of light is recommended to avoid the development of skin inflammation and hyperpigmentation [3].
Alopecia areata is more common in patients with IP [8], most often occurring on the cranial vault, usually secondary to herpes or warty lesions [9]. In addition to the scalp, the trunk and extremities may also be involved [9]. Chan et al. [10] suggested that the pattern of craniovertebral spiral alopecia in patients with IP also corresponds to the Blaschko line on the scalp, i.e. after random X-chromosome inactivation, alopecia occurs in the region of the X chromosome with mutations, and hair is normalized in the region of the X chromosome that does not have mutations.
Nail abnormalities occur mostly after puberty and the most common alterations are in the first three fingers of the hand [9]. The main manifestations are nail bulges [9], indentations [9] and nail dystrophy [1] and in severe cases, nail breakage [1] and subungual keratinized tumors [1]. Advanced warty lesions under the nails require a skin biopsy to confirm the diagnosis, and surgical excision and bone scraping are feasible if the patients feel painful [3].
IP-related retinopathy
Ocular lesions may be seen in 35-77% of patients with IP [11], the most common of which is IP-associated retinopathy (IPR), such as vascular obstruction, neovascularization, vitreoretinal hemorrhage, exudative and tensor retinal detachment [12]. This is followed by optic nerve loss and corneal abnormalities [3]. IPR may involve both eyes or only one eye, and if bilateral involvement is present it tends to be asymmetric in the extent of the lesion [13]. Peng et al. [13] concluded that children with IP who have central nervous system lesions were more likely to develop IPR, and in their retrospective analysis all children with neurologic alterations had ocular involvement, and there was a positive correlation in the severity between neurologic alterations and IPR. Holmström and Thorén [14] first proposed the grading of IPR which has been used since then (Table 1) and in 2019 Peng et al.

[13] conducted a survey on the prevalence of IPR in children with IP in China and updated the guidelines for the grading of IPR (Table 2).

IPR and even retinal detachment can occur as early as 1-week afterbirth [15] and then progresses rapidly, so once the IP is diagnosed, fundus examination should be performed immediately [13]. The multidisciplinary expert consensus on the diagnosis and management of patients with IP, published by the European Network for Rare Skin Diseases in March 2020, states [3] that fundus examination should be performed as soon as IP is diagnosed. If the initial screening result is negative, screening is still required at the following 1, 2, 3, 6, 12, 18, and 24 months and then annually, as a way to rule out the possibility of advanced ocular complications in adult IP patients. Screening recommendations from Chinese experts [13] are as follows: All patients with IP should have a fundus examination by a pediatric ophthalmologist as soon as possible after diagnosis. If the fundus is normal, it should be examined every 3 months for 1 year and twice a year for up to 3 years. They should also be observed for refractive errors and strabismus. After this, patients should be examined every year. FFA should be performed as soon as retinal vasculopathy is detected during the examination or when a normal fundus examination can be used in conjunction with an FFA examination to assess retinal vascular status. Follow-up can be discontinued if all examinations, especially FFA, are normal. However, because IP is a systemic disease, lifelong follow-up at one-year intervals is still strongly recommended.
The treatment of IPR is mainly focused on symptoms, and retinal photocoagulation, intravitreal injection of anti-vascular endothelial growth factor (VEGF) and vitrectomy are often used in clinical practice [16]. Even with early laser therapy, strict post-treatment monitoring is required [15]. Kunzmann et al. reported an adverse event of necrotizing small bowel colitis after an intravitreal bevacizumab injection in a 6-week-old child with IP. They found that the associated systemic uptake of VEGF levels was suppressed after intravitreal injection of bevacizumab, and the suppression of VEGF levels was associated with the development of necrotizing [17]. This study suggests that there may be a link between the development of small bowel colitis and the suppression of VEGF levels [17]. Ranibizumab shorter half-life and a reduced risk of systemic absorption in infants compared with bevacizumab, therefore, Ranibizumab is a preferred choice for treatment. Michel et al. pointed out that anti-VEGF therapy should probably be used as a second-line treatment for severe and atypical cases due to its potential to cause cerebrovascular lesions and stroke in affected infants [15], as well as the relative immaturity of the blood-retinal barrier of infants [15]. The latest report hypothesizes that systemic or topical application of propranolol may alleviate the progression of IPR by blocking neovascularization [18].
IP central nervous system lesions
Central nervous system lesions may be present in 30%-50% of IP patients, mainly manifesting as seizures, microcephaly, mental retardation, and hemiparesis [19]. The pathogenesis of CNS lesions is still not fully understood, and there are two hypotheses: Microvascular lesions leading to vascular occlusions and infarcts; disruption of the immune-regulated nuclear factor kappa-B (NF-κB) pathway leading to increased apoptosis of neuronal and glial cells, or both pathogeneses involved in the damage of the blood-brain barrier disrupting the homeostasis of pro-convulsant factors and inflammation playing a role in the development of small and medium-sized cerebrovascular diseases [20].
Central nervous system lesions usually present in the first week of life [21]. Approximately 30% of neonatal seizures are accompanied by typical cutaneous manifestations [22]. Meanwhile, CNS manifestations are associated with the appearance of the Blistering phase of IP [23]. A case of a 2-year-old patient with IP was reported to present an acute cerebral arteriopathy following influenza A infection, without experiencing a blistering phase in the early stages [19]. The brain MRI of the patient with IP showed multifocal changes, mainly periventricular and cerebral white matter lesions spreading to both hemispheres, and in a few instances, the corpus callosum, basal ganglia, thalamus, or cerebellum were involved [24].
Treatment for CNS lesions is mainly antiepileptic, corticosteroids or adrenocorticosteroids [25] and rehabilitation training. Patients with IP in the neonatal period without neurological damage still require follow-up [3], neurocognitive assessment at 9 and 24 months postnatal, respectively, and MRI of the head at 2 and 3.5 years of age are recommended; when neurological damage is detected, electroencephalogram in the neonatal period, at 4 and 24 months postnatal, respectively, and MRI of the head in the neonatal period and at 30 months is recommended. Regular neurologic and epileptological follow-up is necessary during the disease and throughout life, at least every 6 months within 3 years of age, and systematic neurocognitive assessment after enrollment in elementary school. The frequency of cognitive assessment is determined by the needs of each patient.
IP dental abnormalities
More than 80% of IP patients present dental abnormalities (number, form, size and structure), 18% have delayed eruption, and 40% have some skeletal defects [26]. The most common dental damage is caries, followed by missing teeth, hypodontia, developmental delay, tooth loss and some other oral abnormalities, most commonly cleft palate and high palatal arch [27]. Timely restoration, implantation, and orthodontic rehabilitation are recommended as they follow up with their age [3].
IP breast hypoplasia
Female IP patients may present with excess nipples, nipple hypoplasia, breast hypoplasia and abnormal nipple pigmentation [1].
IP heart lesions
Some cardiac disorders associated with IP have also been reported. Cases of pigment incontinence with ventricular septal defect, atrial septal defect, pulmonary stenosis, and pulmonary hypertension have been reported [21]. Atallah et al. reported a 4-month-old child who developed a collapse during general anesthesia and was subsequently resuscitated and diagnosed with severe pulmonary hypertension [28]. Therefore, once a patient with IP undergoes surgery, the preoperative examinations should include chest radiographs, electrocardiograms, echocardiograms, and a thorough cardiac examination by a cardiologist.
IP susceptibility to autoimmunity, autoinflammation and tumors
IKBKG is a 23-kb gene consisting of 10 exons. Approximately 80% of IP patients are sickened by deletions in exons 4-10 [4]. Non-recurrent genomic rearrangements at the IP locus and point mutations in the IKBKG coding region have also been reported [29]. The NEMO protein, encoded by the IKBKG gene, is responsible for regulating the activity of NF-κB [29]. NF-κB is responsible for the regulation of immune stress response, inflammatory response, cell adhesion and protection against apoptosis [29]. Deletion of the NEMO protein sensitizes the cells to apoptosis, resulting in a male cell
death and female cells selectively biased X inactivation [30]. In addition, the patients with IP are susceptible to encephalitis following viral infections (influenza, herpes, coxsackie and respiratory syncytial virus) and vaccinations (smallpox, diphtheria and tetanus toxoid) [19, 24]. Cerebral arteriolar lesions in patients with IP may be due to the susceptibility of the endothelial cells to proinflammatory cytokines due to mutations in IKBKG [19]. Patients with IP can also develop several different types of cancers [31] and malignancies, such as hematological tumors (acute myeloid leukemia), nephroblastoma, rhabdomyosarcoma and retinoblastoma [32]. Japanese scholars reported a case of basal cell carcinoma occurring in a patient with IP, which may be due to inactivation of the NF-κB signaling pathway as a result of a mutation in the NEMO gene [33]. This mutation may reduce the ability of cells to scavenge reactive oxygen species and prevent the cell cycle from responding to DNA damage, promoting tumor transformation.
IP combined with other rare diseases
Sürmeli Döven reported a case of right polycystic dysplastic kidney and left ureters-bladder junction obstruction with IP without IKBKG/NEMO mutation, and speculated that sequential alignment of the IP-causing genes with other genes involved in renal development could lead to this outcome [34]. Romano et al. reported a case of IP combined with a congenital portal system and nodular regenerative hyperplasia of the liver in a patient with facial dysmorphism and delayed speech, and the genetic results tested positive, which may support the role of NEMO in liver homeostasis [35].
Correlation of IP genotype with phenotype
Phenotypic expression of IKBKG mutations is highly variable [9], phenotypes with the same mutant genotype may be different, whereas the patients with different IKBKG mutations may have the same clinical phenotype [9]. Peng et al. did not find any correlation between genotypes and ocular phenotypes in their study of 61 children with IPR in China [13]. Wang et al. reported a case of an asymptomatic baby boy of IP with a rash and they concluded that somatic mosaicism, a postzygotic mutation occurring at the blastocyst stage of embryogenesis leading to incomplete NF-κB inactivation, occurred in this child, which explained the relatively mild cutaneous symptoms and absence of extracutaneous manifestations in the case [36]. IP patients with negative genetic testing are significantly less likely to have combined dental manifestations [26].
Diagnostic criteria for IP
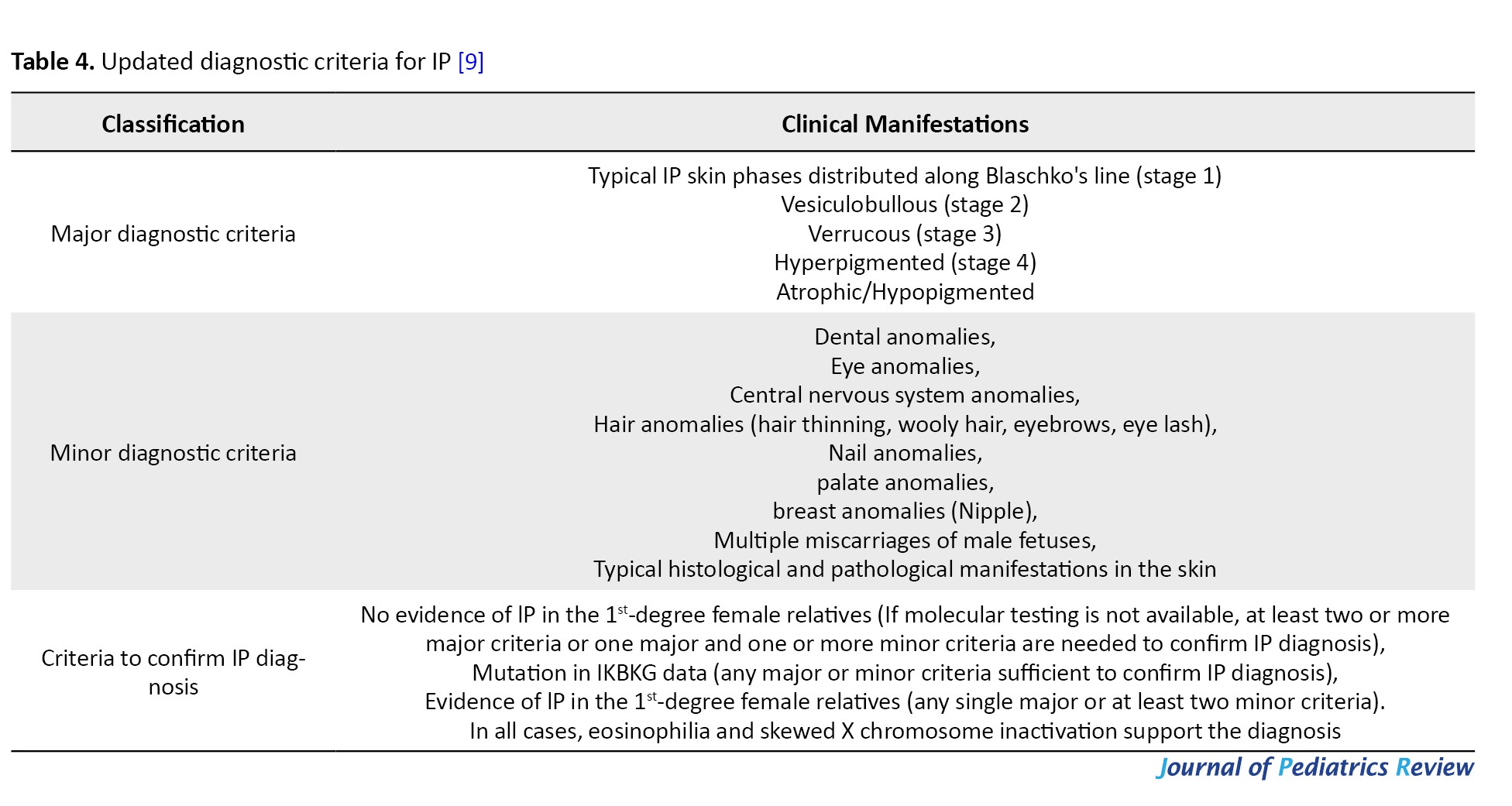
IP was first described by Garrod in 1906 [2], the classic diagnostic criteria for IP were proposed by Landy and Donnai in 1993 [1] (Table 3), and in 2014 the diagnostic criteria for IP were updated to include atypical positive result for mutations in the IKBKG gene and a family history of IP in the diagnostic criteria [9] (Table 4).

The European Network for Rare Skin Diseases in March 2020 published a multidisciplinary expert consensus on the diagnosis and management of patients with IP and also updated the diagnostic criteria [3] (Table 5), emphasizing that in the absence of family history, at least one major criterion is sufficient for the diagnosis of IP.
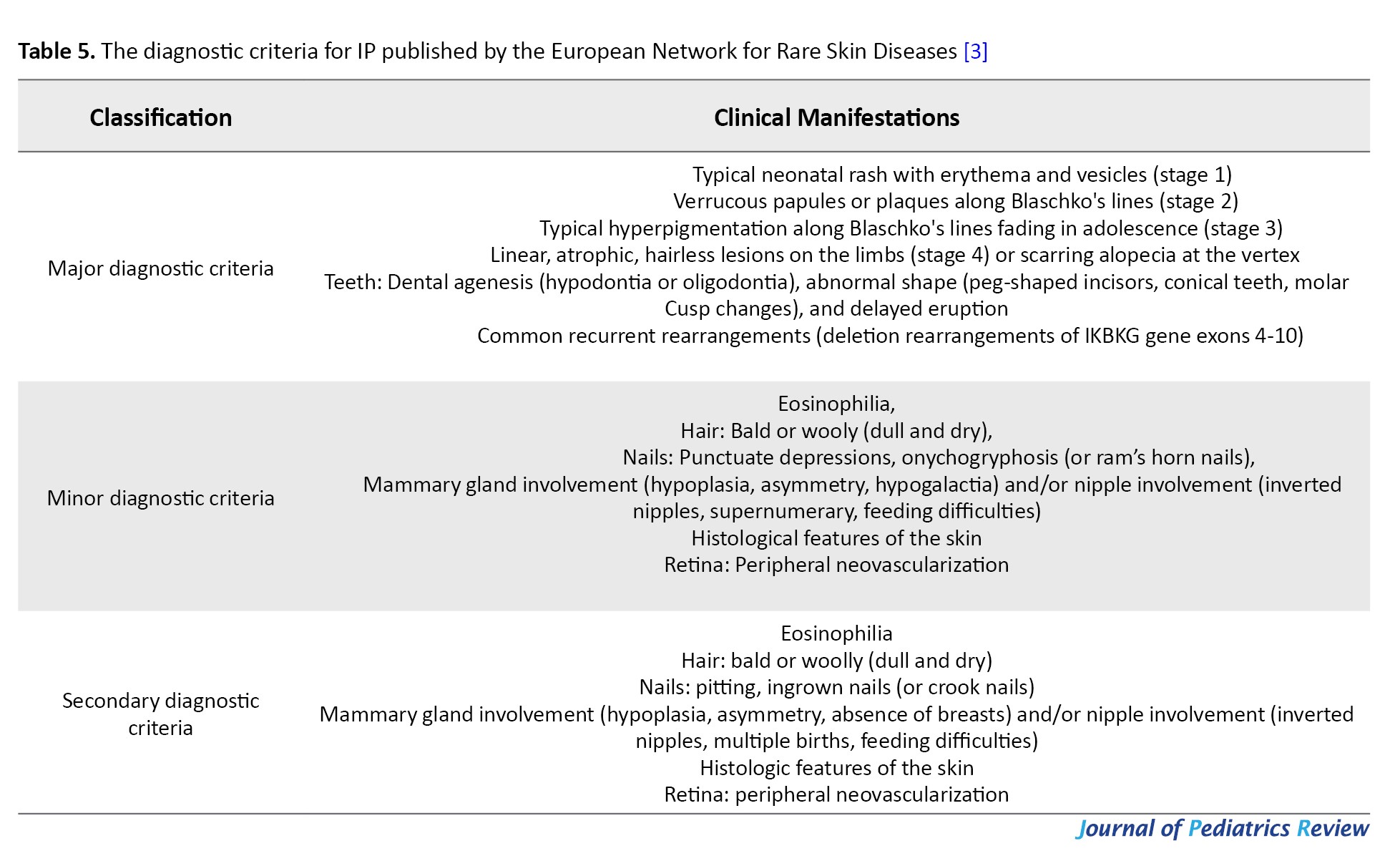
One secondary criterion is sufficient for the diagnosis of IP if IP has been diagnosed in the biological mother. The complete absence of minor criteria should raise some doubt about the diagnosis.
Typical skin manifestations confirm the diagnosis, but older patients in stage IV may require skin biopsy or genetic testing to confirm the diagnosis.
Conclusion
IP is a disease that involves multiple systems and is likely to develop serious ocular and neurologic complications. Once diagnosed, multidisciplinary consultation and evaluation by neurologists, ophthalmologists and dentists are required. Early intervention and adherence to lifelong follow-up are required.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualization, review and editing: Yunfeng Zhang; Data collection, writing the original draf and data analysis: Yuan Wang, Di Ma, Jinpu Zhang and Na Song; Final approval: All authors.
Conflicts of interest
The authors declared no conflict of interest.
References
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, with a prevalence of 0.7-1.2/100 000 [1, 2], is an X-chromosome-linked dominant disorder caused by mutations in the inhibitor of nuclear factor kappa-B kinase subunit gamma (NEMO/IKBKG) gene mutation at Xq28 [1]. More than 75% of IP patients are sporadic cases and the rest are familial about 10-25% [3]. It is usually fatal for male fetuses, mostly dying in utero [1]. In rare cases, male IP patients can survive if combined with Klinefelter syndrome (47, XXY karyotype), subtype mutations, or IKBKG somatic mosaicism [1, 2, 4, 5].
Case Presentation
Case number 1
A female patient was admitted to our hospital with a postnatal rash. The patient was delivered by cesarean section at a gestational age of 39 weeks with a birth weight of 3600 g. A family history of skin condition was denied. The mother had a 13-year-old daughter with no history of rash. After birth, the patient was found to have scattered pustules all over the skin, which protruded from the skin surface, with no obvious ulceration or exudation. Blood routine test results showed a leukocyte count of 40.8×109/L, a neutrophil percentage of 76.5%, a neutrophil count of 31.20×109/L, an eosinophil percentage of 7.3% and an eosinophil count of 1.40×109/L. The patient was administered cefepime for anti-infective treatment. On the second day of admission, most of the generalized rash subsided, leaving hyperpigmentation and dandruff and scattered herpes was still seen on the limbs, and the routine blood test results showed that the
white blood cell count was 20.6×109/L, the percentage of neutrophil was 58.2%, the neutrophil count was 12.01×109/L, the percentage of eosinophils was 2.04%, and the count of eosinophils was 0.10×109/L. On the fifth day of admission, red papules appeared on the limbs, and after consultation with the doctor from the Department of Dermatology, the diagnosis of IP was assigned. Systemic application of antibiotics was discontinued and topical skin healing film (2-3 times/day) was administered to prevent skin infections. Fundus examination and fundus fluorescein angiography (FFA) showed ischemic changes in the retina of both eyes (Figure 1 A-D), and retinal laser photocoagulation was performed in both eyes.
Case number 2
A 4-month-old female infant was diagnosed with IP on skin biopsy at another hospital (Figure 2A).
Case number 3
A female infant, presented to our department 20 days after birth with a rash (Figure 1F), whose mother was an IP patient with a history of miscarriage of male fetus. A complete head MRI of fundus cardiac color showed no abnormality. She is currently under regular follow-up.
Discussion
IP is an ectodermal disease that involves multiple systems and is characterized by skin pigmentation accompanied by damage to the teeth, eyes, central nervous system, mammary glands, and bones [1]. The following is an overview of the presentation and treatment of IP for each system.
IP skin performance
Skin changes are the most typical manifestations of IP, and the diagnosis can be confirmed based on skin manifestations alone [1]. The cutaneous manifestations occur sequentially in four stages, not all of which are mandatory and may overlap. The first stage is characterized by blisters distributed along Blaschko’s line, which most often occur in the neonatal period and may be accompanied by an elevated peripheral blood leukocyte count and eosinophil count. The rash may recur during febrile illnesses, infections (coxsackievirus) and vaccinations [6]. In the second stage, the blisters heal and form warty lesions and hyperkeratosis. The third stage is the hyperpigmentation phase, which can last until adulthood. The last stage mostly occurs before the complete disappearance of hyperpigmentation and is characterized by pale, hairless lesions [1].
Hadj-Rabia et al. [7] described the pathohistological changes in the skin at each stage and proposed them as secondary criteria for IP. Adults with IP may no longer have typical skin manifestations [8]. The manifestations of gray-blue mottled alopecia without capillary dilatation and follicular exfoliation/clogging found on dermoscopy can help in the diagnosis of IP. Skin damage usually resolves spontaneously without treatment, and in a few severe cases, it can be treated with topical hormonal therapy [3]. Both case 1 and case 2 reported in this paper had skin lesions combined with skin infections, which gradually healed after administration of systemic anti-infection therapy. In neonates, topical sterile dressings can be applied to prevent infections [3], and the skin healing film applied in case 1 is a sterile dressing, which achieved good results in case 1. Laser treatment can be attempted for recurrent inflammation, while avoidance of light is recommended to avoid the development of skin inflammation and hyperpigmentation [3].
Alopecia areata is more common in patients with IP [8], most often occurring on the cranial vault, usually secondary to herpes or warty lesions [9]. In addition to the scalp, the trunk and extremities may also be involved [9]. Chan et al. [10] suggested that the pattern of craniovertebral spiral alopecia in patients with IP also corresponds to the Blaschko line on the scalp, i.e. after random X-chromosome inactivation, alopecia occurs in the region of the X chromosome with mutations, and hair is normalized in the region of the X chromosome that does not have mutations.
Nail abnormalities occur mostly after puberty and the most common alterations are in the first three fingers of the hand [9]. The main manifestations are nail bulges [9], indentations [9] and nail dystrophy [1] and in severe cases, nail breakage [1] and subungual keratinized tumors [1]. Advanced warty lesions under the nails require a skin biopsy to confirm the diagnosis, and surgical excision and bone scraping are feasible if the patients feel painful [3].
IP-related retinopathy
Ocular lesions may be seen in 35-77% of patients with IP [11], the most common of which is IP-associated retinopathy (IPR), such as vascular obstruction, neovascularization, vitreoretinal hemorrhage, exudative and tensor retinal detachment [12]. This is followed by optic nerve loss and corneal abnormalities [3]. IPR may involve both eyes or only one eye, and if bilateral involvement is present it tends to be asymmetric in the extent of the lesion [13]. Peng et al. [13] concluded that children with IP who have central nervous system lesions were more likely to develop IPR, and in their retrospective analysis all children with neurologic alterations had ocular involvement, and there was a positive correlation in the severity between neurologic alterations and IPR. Holmström and Thorén [14] first proposed the grading of IPR which has been used since then (Table 1) and in 2019 Peng et al.
[13] conducted a survey on the prevalence of IPR in children with IP in China and updated the guidelines for the grading of IPR (Table 2).
IPR and even retinal detachment can occur as early as 1-week afterbirth [15] and then progresses rapidly, so once the IP is diagnosed, fundus examination should be performed immediately [13]. The multidisciplinary expert consensus on the diagnosis and management of patients with IP, published by the European Network for Rare Skin Diseases in March 2020, states [3] that fundus examination should be performed as soon as IP is diagnosed. If the initial screening result is negative, screening is still required at the following 1, 2, 3, 6, 12, 18, and 24 months and then annually, as a way to rule out the possibility of advanced ocular complications in adult IP patients. Screening recommendations from Chinese experts [13] are as follows: All patients with IP should have a fundus examination by a pediatric ophthalmologist as soon as possible after diagnosis. If the fundus is normal, it should be examined every 3 months for 1 year and twice a year for up to 3 years. They should also be observed for refractive errors and strabismus. After this, patients should be examined every year. FFA should be performed as soon as retinal vasculopathy is detected during the examination or when a normal fundus examination can be used in conjunction with an FFA examination to assess retinal vascular status. Follow-up can be discontinued if all examinations, especially FFA, are normal. However, because IP is a systemic disease, lifelong follow-up at one-year intervals is still strongly recommended.
The treatment of IPR is mainly focused on symptoms, and retinal photocoagulation, intravitreal injection of anti-vascular endothelial growth factor (VEGF) and vitrectomy are often used in clinical practice [16]. Even with early laser therapy, strict post-treatment monitoring is required [15]. Kunzmann et al. reported an adverse event of necrotizing small bowel colitis after an intravitreal bevacizumab injection in a 6-week-old child with IP. They found that the associated systemic uptake of VEGF levels was suppressed after intravitreal injection of bevacizumab, and the suppression of VEGF levels was associated with the development of necrotizing [17]. This study suggests that there may be a link between the development of small bowel colitis and the suppression of VEGF levels [17]. Ranibizumab shorter half-life and a reduced risk of systemic absorption in infants compared with bevacizumab, therefore, Ranibizumab is a preferred choice for treatment. Michel et al. pointed out that anti-VEGF therapy should probably be used as a second-line treatment for severe and atypical cases due to its potential to cause cerebrovascular lesions and stroke in affected infants [15], as well as the relative immaturity of the blood-retinal barrier of infants [15]. The latest report hypothesizes that systemic or topical application of propranolol may alleviate the progression of IPR by blocking neovascularization [18].
IP central nervous system lesions
Central nervous system lesions may be present in 30%-50% of IP patients, mainly manifesting as seizures, microcephaly, mental retardation, and hemiparesis [19]. The pathogenesis of CNS lesions is still not fully understood, and there are two hypotheses: Microvascular lesions leading to vascular occlusions and infarcts; disruption of the immune-regulated nuclear factor kappa-B (NF-κB) pathway leading to increased apoptosis of neuronal and glial cells, or both pathogeneses involved in the damage of the blood-brain barrier disrupting the homeostasis of pro-convulsant factors and inflammation playing a role in the development of small and medium-sized cerebrovascular diseases [20].
Central nervous system lesions usually present in the first week of life [21]. Approximately 30% of neonatal seizures are accompanied by typical cutaneous manifestations [22]. Meanwhile, CNS manifestations are associated with the appearance of the Blistering phase of IP [23]. A case of a 2-year-old patient with IP was reported to present an acute cerebral arteriopathy following influenza A infection, without experiencing a blistering phase in the early stages [19]. The brain MRI of the patient with IP showed multifocal changes, mainly periventricular and cerebral white matter lesions spreading to both hemispheres, and in a few instances, the corpus callosum, basal ganglia, thalamus, or cerebellum were involved [24].
Treatment for CNS lesions is mainly antiepileptic, corticosteroids or adrenocorticosteroids [25] and rehabilitation training. Patients with IP in the neonatal period without neurological damage still require follow-up [3], neurocognitive assessment at 9 and 24 months postnatal, respectively, and MRI of the head at 2 and 3.5 years of age are recommended; when neurological damage is detected, electroencephalogram in the neonatal period, at 4 and 24 months postnatal, respectively, and MRI of the head in the neonatal period and at 30 months is recommended. Regular neurologic and epileptological follow-up is necessary during the disease and throughout life, at least every 6 months within 3 years of age, and systematic neurocognitive assessment after enrollment in elementary school. The frequency of cognitive assessment is determined by the needs of each patient.
IP dental abnormalities
More than 80% of IP patients present dental abnormalities (number, form, size and structure), 18% have delayed eruption, and 40% have some skeletal defects [26]. The most common dental damage is caries, followed by missing teeth, hypodontia, developmental delay, tooth loss and some other oral abnormalities, most commonly cleft palate and high palatal arch [27]. Timely restoration, implantation, and orthodontic rehabilitation are recommended as they follow up with their age [3].
IP breast hypoplasia
Female IP patients may present with excess nipples, nipple hypoplasia, breast hypoplasia and abnormal nipple pigmentation [1].
IP heart lesions
Some cardiac disorders associated with IP have also been reported. Cases of pigment incontinence with ventricular septal defect, atrial septal defect, pulmonary stenosis, and pulmonary hypertension have been reported [21]. Atallah et al. reported a 4-month-old child who developed a collapse during general anesthesia and was subsequently resuscitated and diagnosed with severe pulmonary hypertension [28]. Therefore, once a patient with IP undergoes surgery, the preoperative examinations should include chest radiographs, electrocardiograms, echocardiograms, and a thorough cardiac examination by a cardiologist.
IP susceptibility to autoimmunity, autoinflammation and tumors
IKBKG is a 23-kb gene consisting of 10 exons. Approximately 80% of IP patients are sickened by deletions in exons 4-10 [4]. Non-recurrent genomic rearrangements at the IP locus and point mutations in the IKBKG coding region have also been reported [29]. The NEMO protein, encoded by the IKBKG gene, is responsible for regulating the activity of NF-κB [29]. NF-κB is responsible for the regulation of immune stress response, inflammatory response, cell adhesion and protection against apoptosis [29]. Deletion of the NEMO protein sensitizes the cells to apoptosis, resulting in a male cell
death and female cells selectively biased X inactivation [30]. In addition, the patients with IP are susceptible to encephalitis following viral infections (influenza, herpes, coxsackie and respiratory syncytial virus) and vaccinations (smallpox, diphtheria and tetanus toxoid) [19, 24]. Cerebral arteriolar lesions in patients with IP may be due to the susceptibility of the endothelial cells to proinflammatory cytokines due to mutations in IKBKG [19]. Patients with IP can also develop several different types of cancers [31] and malignancies, such as hematological tumors (acute myeloid leukemia), nephroblastoma, rhabdomyosarcoma and retinoblastoma [32]. Japanese scholars reported a case of basal cell carcinoma occurring in a patient with IP, which may be due to inactivation of the NF-κB signaling pathway as a result of a mutation in the NEMO gene [33]. This mutation may reduce the ability of cells to scavenge reactive oxygen species and prevent the cell cycle from responding to DNA damage, promoting tumor transformation.
IP combined with other rare diseases
Sürmeli Döven reported a case of right polycystic dysplastic kidney and left ureters-bladder junction obstruction with IP without IKBKG/NEMO mutation, and speculated that sequential alignment of the IP-causing genes with other genes involved in renal development could lead to this outcome [34]. Romano et al. reported a case of IP combined with a congenital portal system and nodular regenerative hyperplasia of the liver in a patient with facial dysmorphism and delayed speech, and the genetic results tested positive, which may support the role of NEMO in liver homeostasis [35].
Correlation of IP genotype with phenotype
Phenotypic expression of IKBKG mutations is highly variable [9], phenotypes with the same mutant genotype may be different, whereas the patients with different IKBKG mutations may have the same clinical phenotype [9]. Peng et al. did not find any correlation between genotypes and ocular phenotypes in their study of 61 children with IPR in China [13]. Wang et al. reported a case of an asymptomatic baby boy of IP with a rash and they concluded that somatic mosaicism, a postzygotic mutation occurring at the blastocyst stage of embryogenesis leading to incomplete NF-κB inactivation, occurred in this child, which explained the relatively mild cutaneous symptoms and absence of extracutaneous manifestations in the case [36]. IP patients with negative genetic testing are significantly less likely to have combined dental manifestations [26].
Diagnostic criteria for IP
IP was first described by Garrod in 1906 [2], the classic diagnostic criteria for IP were proposed by Landy and Donnai in 1993 [1] (Table 3), and in 2014 the diagnostic criteria for IP were updated to include atypical positive result for mutations in the IKBKG gene and a family history of IP in the diagnostic criteria [9] (Table 4).
The European Network for Rare Skin Diseases in March 2020 published a multidisciplinary expert consensus on the diagnosis and management of patients with IP and also updated the diagnostic criteria [3] (Table 5), emphasizing that in the absence of family history, at least one major criterion is sufficient for the diagnosis of IP.
One secondary criterion is sufficient for the diagnosis of IP if IP has been diagnosed in the biological mother. The complete absence of minor criteria should raise some doubt about the diagnosis.
Typical skin manifestations confirm the diagnosis, but older patients in stage IV may require skin biopsy or genetic testing to confirm the diagnosis.
Conclusion
IP is a disease that involves multiple systems and is likely to develop serious ocular and neurologic complications. Once diagnosed, multidisciplinary consultation and evaluation by neurologists, ophthalmologists and dentists are required. Early intervention and adherence to lifelong follow-up are required.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualization, review and editing: Yunfeng Zhang; Data collection, writing the original draf and data analysis: Yuan Wang, Di Ma, Jinpu Zhang and Na Song; Final approval: All authors.
Conflicts of interest
The authors declared no conflict of interest.
References
- Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993; 30(1):53-9. [DOI:10.1136/jmg.30.1.53] [PMID]
- HaqueMN, Ohtsubo M, Nishina S, Nakao S, Yoshida K, Hosono K, et al. Analysis of IKBKG/NEMO gene in five Japanese cases of incontinentia pigmenti with retinopathy: Fine genomic assay of a rare male case with mosaicism. J Hum Genet. 2021; 66(6):205-14. [DOI:10.1038/s10038-021-00900-6] [PMID]
- Bodemer C, Diociaiuti A, Hadj-Rabia S, Robert MP, Desguerre I, Manière MC, et al. Multidisciplinary consensus recommendations from a European network for the diagnosis and practical management of patients withincontinentia pigmenti. J Eur Acad Dermatol Venereol. 2020; 34(7):1415-24. [DOI:10.1111/jdv.16403] [PMID]
- Shibata K, Kunisada M, Miyai S, Kawamori S, Kurahashi H, Nishigori C. Incontinentia pigmenti in a female infant with somatic mosaicism due to the IKBKG variant. J Dermatol. 2021; 48(12):e577-8. [DOI:10.1111/1346-8138.16141]
- Thorsness S, Eyler J, Mudaliar K, SpeiserJ, Kim W. Asymptomatic Rash in a male infant with incontinentia pigmenti. J Pediatr. 2019; 215:278-278.e1. [DOI:10.1016/j.jpeds.2019.07.005] [PMID]
- Moro R, Fabiano A, Calzavara-Pinton P, Cardinale J, Palumbo G, Giliani S, et al. Incontinentia pigmenti associated with aplasia cutis congenita in a newborn male with klinefelter syndrome: Is the severity of neurological involvement linked to skin manifestations? Dermatol Ther (Heidelb). 2020; 10(1):213-20. [DOI:10.1007/s13555-019-00336-z] [PMID]
- Hadj-Rabia S, Rimella A, Smahi A, Fraitag S, Hamel-TeillacD, Bonnefont JP, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κB essential modulator gene mutations. J Am Acad Dermatol. 2011; 64(3):508-15. [DOI:10.1016/j.jaad.2010.01.045] [PMID]
- Razmi T M, Jogunoori S, Radotra BD, De D. Trichoscopy of whorled alopecia revealing “pigment incontinence” of incontinentia pigmenti. Int J Dermatol. 2019; 58(8):e156-8. [DOI:10.1111/ijd.14481] [PMID]
- Minić S, TrpinacD, Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014; 85(6):536-42. [DOI:10.1111/cge.12223] [PMID]
- Chan YC, Happle R, Giam YC. Whorled scarring alopecia: A rare phenomenon in incontinentia pigmenti? J Am Acad Dermatol. 2003; 49(5):929-31. [DOI:10.1016/S0190-9622(03)00474-2] [PMID]
- Jiang JJ, Zhang Q, Ding ZX, Qiu MY. Fundus manifestations and treatment of pigment incontinence. Int Eye Sci. 2021, 21(03):561-6. [DOI: 10.3980/j.issn.1672-5123.2021.3.39]
- Nakao S, Nishina S, Tanaka S, Yoshida T, Yokoi T, Azuma N. Early laser photocoagulation for extensive retinal avascularity in infants with incontinentia pigmenti. Jpn J Ophthalmol. 2020; 64(6):613-20. [DOI:10.1007/s10384-020-00768-7] [PMID]
- Peng J, Zhang Q, Long X, Zhang J, Huang Q, Li Y, et al. Incontinentia pigmenti- associated ocular anomalies of paediatric incontinentia pigmenti patients in China. Acta Ophthalmol. 2019; 97(3):265-72. [DOI:10.1111/aos.13781] [PMID]
- Holmström G, Thorén K. Ocular manifestations of incontinentia pigmenti. Acta Ophthalmol Scand. 2000; 78(3):348-53. [DOI:10.1034/j.1600-0420.2000.078003348.x] [PMID]
- Michel S, Reynaud C, Daruich A, Hadj-Rabia S, Bremond-Gignac D, Bodemer C, et al. Early management of sight threatening retinopathy in incontinentia pigmenti. Orphanet J Rare Dis. 2020; 15(1):223. [DOI:10.1186/s13023-020-01509-2] [PMID]
- Yin Y. Anti-VEGF therapy in pediatric retinal and choroidal disease. Int Eye Sci. 2021, 12:85-8. [Link]
- Kunzmann S, Ngyuen T, Stahl A, Walz JM, Nentwich MM, Speer CP, et al. Necrotizing enterocolitis after intravitreal bevacizumab in an infant with Incontinentia Pigmenti - a case report. BMC Pediatr. 2019; 19(1):353. [DOI:10.1186/s12887-019-1732-z] [PMID]
- Oranges T, El HachemM, Filippeschi C, Romanelli M, Filippi L. The potential role of propranolol in incontinentia pigmenti. Dermatol Ther. 2021; 34(1):e14737. [DOI:10.1111/dth.14737]
- Kanai S, Okanishi T, Kawai M, Yoshino G, Tsubouchi Y, Nishimura Y, et al. Late- onset cerebral arteriopathy in a patient with incontinentia pigmenti. Brain Dev. 2021; 43(4):580-4. [DOI:10.1016/j.braindev.2020.12.015] [PMID]
- Kim HY, Song HB, Kim KH, Kim JH, Chae JH, Kim MJ, et al. Importance of extracutaneous organ involvement in determining the clinical severity and prognosis of incontinentia pigmenti caused by mutations in the IKBKG gene. Exp Dermatol. 2021; 30(5):676-83. [DOI:10.1111/exd.14313] [PMID]
- Sugur T, Kavakli AS, Metinyurt HF. Anaesthesia and orphan disease: A child with incontinentia pigmenti. Eur J Anaesthesiol. 2020; 37(2):141-3. [DOI:10.1097/EJA.0000000000001136] [PMID]
- Minić S, Trpinac D, Obradović M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013; 8:25. [DOI:10.1186/1750-1172-8-25] [PMID]
- Peebles JK, Craddock L, Bennett DD, Longley BJ, Swanson A. Incontinentia Pigmenti: Initial presentation of encephalopathy and seizures. Cutis. 2021; 107(3):E37-9. [DOI:10.12788/cutis.0227] [PMID]
- Rr P, Douch C, Aan Koh MJ, Lai AHM, Lim CT, Hartley L, et al. Speckled brain lesions in Incontinentia Pigmenti patients with acquired brain syndromes. Eur J Paediatr Neurol. 2021; 33:106-11. [DOI:10.1016/j.ejpn.2021.05.012] [PMID]
- Sharawat IK, Panda PK. Epileptic spasms in an infant withincontinentia pigmenti: Report of a rare case with brief review of the literature. J Neurosci Rural Pract. 2020; 11(2):325-8. [DOI:10.1055/s-0040-1709246] [PMID]
- Santa Maria FD, Barros SE, Chiqueto K, Mariath LM, Schüler-Faccini L, Kiszewski AE. Development of dentofacial characteristics related to Incontinentia Pigmenti syndrome: A repeated cross-sectional study. Am J Orthod Dentofacial Orthop. 2021; 160(1):66-76. [DOI:10.1016/j.ajodo.2020.03.033] [PMID]
- Ocaña Jaramillo S, Del Boz J, Vera Casaño Á. [Incontinentia pigmenti. A descriptive study of experience in two different hospitals (Spanish)]. An Pediatr (Engl Ed). 2020; 92(1):3-12. [DOI:10.1016/j.anpedi.2019.04.004] [PMID]
- Atallah V, Meot M, Kossorotoff M, Galmiche-Rolland L, Lardeux C, Neven B, Bodemer C, Bonnet D. A case of reversible pulmonary arterial hypertension associated with incontinentia pigmenti. Pulm Circ. 2018; 8(4):2045894018793983. [DOI: 10.1177/2045894018793983] [PMID]
- Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. 2000; 405(6785):466-72. [PMID]
- Smahi A, Courtois G, Rabia SH, Döffinger R, Bodemer C, Munnich A, et al. The NF-kappaB signalling pathway in human diseases: From incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002; 11(20):2371-5. [DOI:10.1093/hmg/11.20.2371] [PMID]
- Gibson DC, Couser NL, King KB. Co-occurrence of incontinentia pigmenti and down syndrome: Examining patients’ potential susceptibility to autoimmune disease, autoinflammatory disease, cancer, and significant ocular disease. Ophthalmic Genet. 2021; 42(1):92-5. [DOI:10.1080/13816810.2020.1839917] [PMID]
- Tandon S, Prasad M, Vora T, Chinnaswamy G, Shetye N. A rare association of retinoblastoma with incontinentia pigmenti. J Pediatr Hematol Oncol. 2020; 42(5):372-4. [DOI:10.1097/MPH.0000000000001797] [PMID]
- Coppola R, Devirgiliis V, Carbotti M, Zanframundo S, Roberti V, Panasiti V. A case of basal cell carcinoma in a young patient withincontinentia pigmenti. G Ital Dermatol Venereol. 2020; 155(4):526-7. [DOI:10.23736/S0392-0488.18.06081-9]
- Sürmeli Döven S, Delibas A, Türsen Ü, Ezgü FS. Multicystic dysplastic kidney and incontinentia pigmenti: Coexistence of 2 rare diseases. Iran J Kidney Dis. 2019; 13(1):67-70. [PMID]
- RomanoR, Grasso F, Gallo V, CirilloE, Prencipe R, Mamone G, et al. A case of incontinentiapigmenti associated with congenital absence of portal vein system and nodular regenerative hyperplasia. Br J Dermatol. 2019; 180(3):674-5. [DOI:10.1111/bjd.17319] [PMID]
- Wang GL, Deng XL, Peng J, Wang X, Wu LW, Zhang CL et al. Diagnosis and treatment of 12 cases of febrile infection-related epilepsy syndrome. Chin Physician J. 2019, 21(9):1297-301. [Link]
Type of Study: Case Report and Review of Literature |
Subject:
Pediatrics
Received: 2024/05/25 | Accepted: 2024/06/23 | Published: 2024/10/1
Received: 2024/05/25 | Accepted: 2024/06/23 | Published: 2024/10/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
