
Sun, Jun 14, 2026


Volume 13, Issue 2 (4-2025)
J. Pediatr. Rev 2025, 13(2): 125-132 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Lashkarbolouk N, Mazandarani M, Ahani Azari A, Shahkar L. Examining the Clinical Insights Into the Diagnosis of Systemic Lupus Erythematosus in a 7-year-old Patient: A Case Report and Literature Review. J. Pediatr. Rev 2025; 13 (2) :125-132
URL: http://jpr.mazums.ac.ir/article-1-660-en.html
URL: http://jpr.mazums.ac.ir/article-1-660-en.html



1- Taleghani Pediatric Hospital, Golestan University of Medical Sciences, Gorgan, Iran. & Endocrinology and Metabolism Research Institute, Tehran University of Medical Sciences, Tehran, Iran.
2- Taleghani Pediatric Hospital, Golestan University of Medical Sciences, Gorgan, Iran.
3- Taleghani Pediatric Hospital, Golestan University of Medical Sciences, Gorgan, Iran. ,lobatshahkar@yahoo.com
2- Taleghani Pediatric Hospital, Golestan University of Medical Sciences, Gorgan, Iran.
3- Taleghani Pediatric Hospital, Golestan University of Medical Sciences, Gorgan, Iran. ,
Keywords: Hypocomplementemia, Complement C4, Complement C3, Systemic lupus erythematosus (SLE), Pediatrics
Full-Text [PDF 737 kb]
(905 Downloads)
| Abstract (HTML) (1898 Views)
Full-Text: (388 Views)
Introduction
Systemic lupus erythematosus (SLE) is a prototype of a multi-system autoimmune disease that arises from a complex etiopathogenesis. Typically, the disease affects the skin, the musculoskeletal system, and the kidneys. The exact cause and mechanism of SLE still need to be fully understood. Still, it is considered to involve a combination of genetic, environmental, and hormonal factors [1, 2].
In children, unusual clinical presentation with uncommon symptoms happens, mainly at a young age (under six years old), and should be considered a chronological history of the family history of autoimmunity and all relevant events. The disease often manifests with signs affecting multiple organ systems in children and adolescents [1, 2]. Early-onset SLE is linked to specific complement deficiencies in the classical pathway (C1, C4, and C2), which may increase susceptibility to SLE at a young age. These deficiencies can contribute to the dysregulation of the immune system, leading to the production of autoantibodies and the development of systemic inflammation characteristic of SLE [3].
This chronic disease is characterized by variability in clinical phenotypes and the production of multiple autoantibodies. The disease presents with a wide range of symptoms and signs, including photosensitivity rash, hair loss, arthritis, arthralgia, hematuria, proteinuria, hypergammaglobulinemia, hypocomplementemia, and positive rheumatological tests. SLE patients may display acute and severe disease with seizure, psychosis, uremia, pancreatitis, pulmonary hemorrhage, or sepsis [3].
The diagnosis of SLE is determined based on a range of clinical symptoms, serological and laboratory tests, biopsies, or other diagnostic procedures. Key diagnostic tools include blood tests for specific antibodies (such as antinuclear antibodies [ANA], anti-double-stranded DNA [anti-dsDNA], and anti-Smith antibody), with elevated levels indicating SLE. Urinalysis can reveal kidney involvement by detecting protein or blood, while imaging techniques like ultrasound, x-rays, or computed tomography scans may assess organ involvement, particularly in the kidneys and lungs [4, 5].
Early diagnosis and treatment are crucial for managing the disease and preventing organ damage. The diagnosis of SLE can be complex, and if SLE is suspected, a comprehensive evaluation and ongoing monitoring are typically needed to confirm the diagnosis and develop an appropriate treatment plan.
Case Presentation
A 7-year-old boy Baloch patient with a past medical history of C3 and C4 hypocomplementemia, was referred to our pediatric hospital with a chief complaint of new skin lesions. He was taking prednisolone, alendronate, and calcium supplements. He had been treated with the rituximab protocol, which was discontinued about six months ago. The lesions have gradually appeared and have intensified over the past month. The lesions were not itchy, and there was no discharge. The patient also reports that the lesions worsened after exposure to light. Moreover, he suffered from arthralgia and bone pain. There was no history of fever, dyspnea, chest pain, or neurological symptoms.
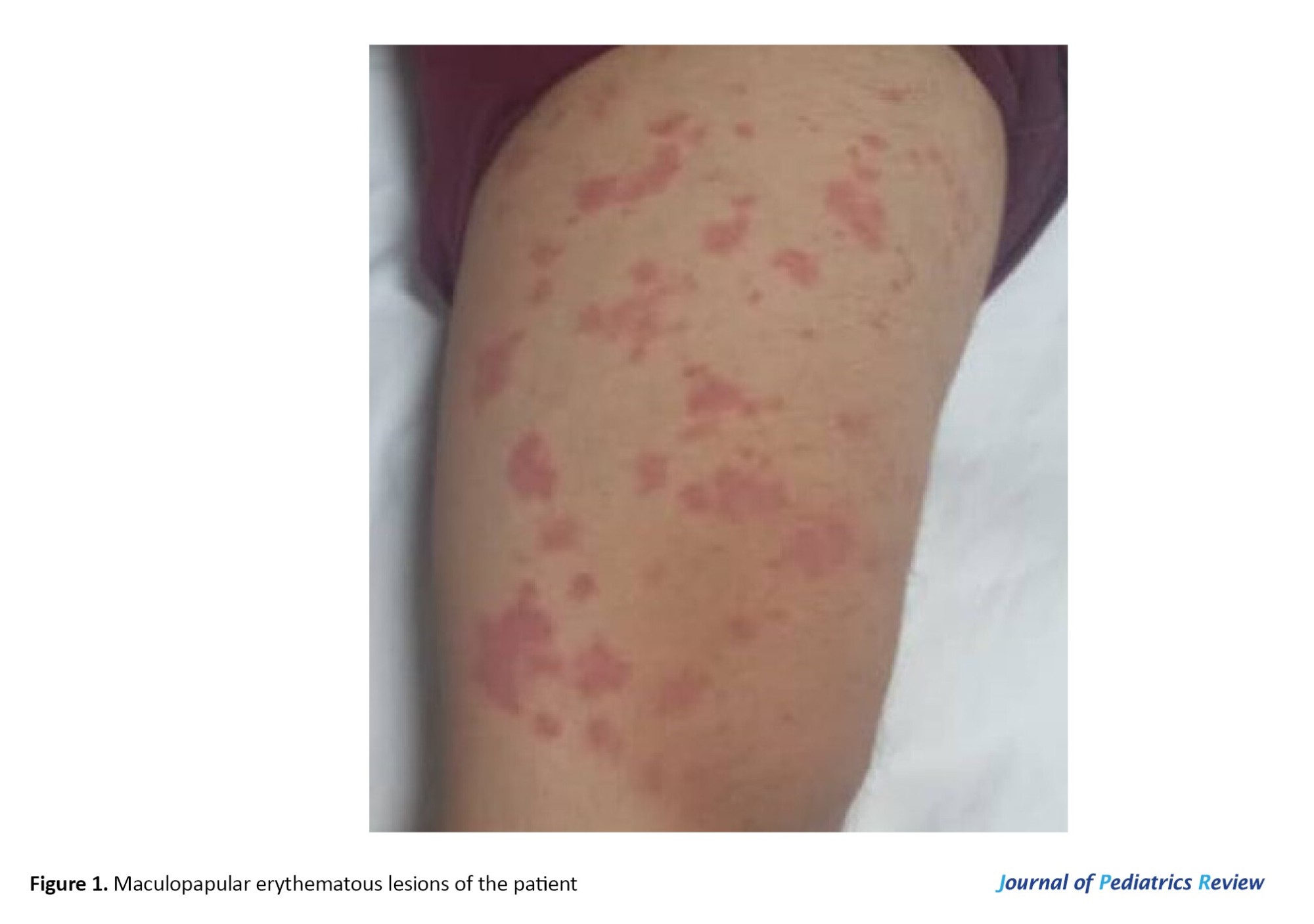
In his physical examination, scattered maculopapular erythematous and scaling lesions were observed in both upper and lower limbs (Figure 1). His vital signs were stable and neurological, and fundus examinations were intact. There was no sign or history of malar rash, arthritis, hepatosplenomegaly, or lymphadenopathy. According to the patient’s parents, due to improvement in their child’s clinical condition and normal complement level, they discontinued rituximab treatment about six months ago.
In the maternal lineage, there were no issues during pregnancy and childbirth, and no birth defects were noted in the individual’s siblings. There was no familial background of genetic susceptibility or hereditary cancer-related conditions. Additionally, his parents do not relate.
In his past medical history, he had a history of bone pain, arthralgia, and intermittent fever when he was 2 years old. Due to the presence of anemia and normal rheumatologic laboratory tests, he was diagnosed with juvenile idiopathic arthritis (JRA) and was treated with methotrexate along with oral corticosteroids. Later, he had an episode of severe dyspnea, low oxygen saturation level, and hemoptysis when he was three years old. He was admitted with an initial diagnosis of diffuse alveolar hemorrhage (DAH). During his hospitalization, there were high levels of C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), anemia, and hypocomplementemia (low C4 and C3). Normal ANA, anti-dsDNA, rheumatoid factor (RF), anti-cyclic citrullinated peptide (anti-CCP), anti-neutrophil cytoplasmic Autoantibody, Cytoplasmic (C-ANCA), perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA), anti-Ro, anti-La, lupus anticoagulant, anti-glomerular basement membrane diseases (Anti-GBM), anti-cardiolipin Ab, anti-phospholipid Ab, and urine analysis were also observed. The diagnosis of hypocomplementemia was confirmed for him. When he was 4 years old, he also had a history of severe pulmonary cytomegalovirus (CMV) infection, which was treated with ganciclovir.
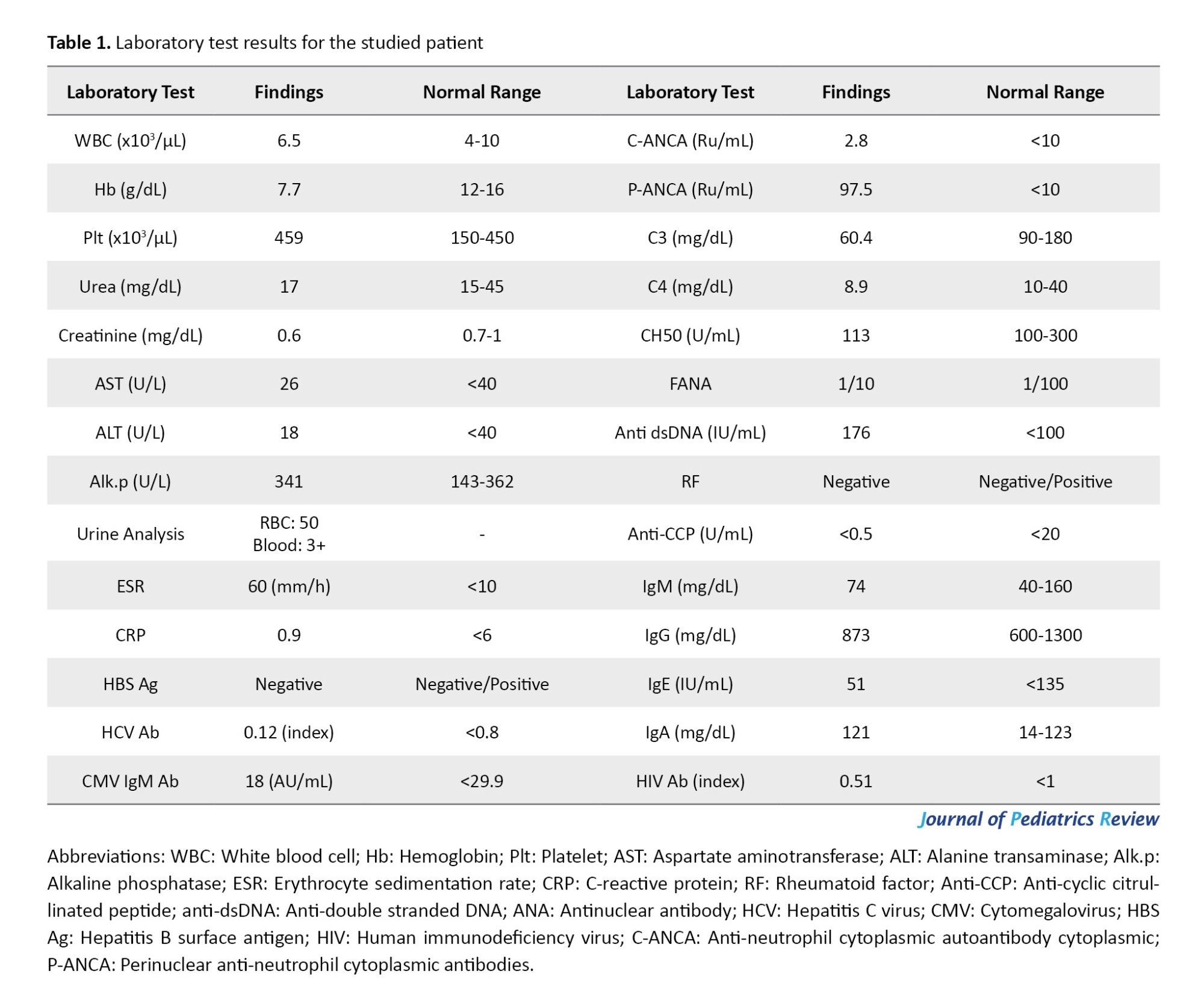
In our evaluation, he had anemia, high ESR level, hematuria, low levels of C3 and C4, elevated P-ANCA level, and positive anti-dsDNA with a normal amount of other rheumatological tests. The viral markers were negative for human immunodeficiency virus (HIV), CMV, hepatitis B virus, and hepatitis C virus (HCV). The immunoglobulin (Ig) M, IgG, IgE, and IgA levels were also in the normal range (Table 1).
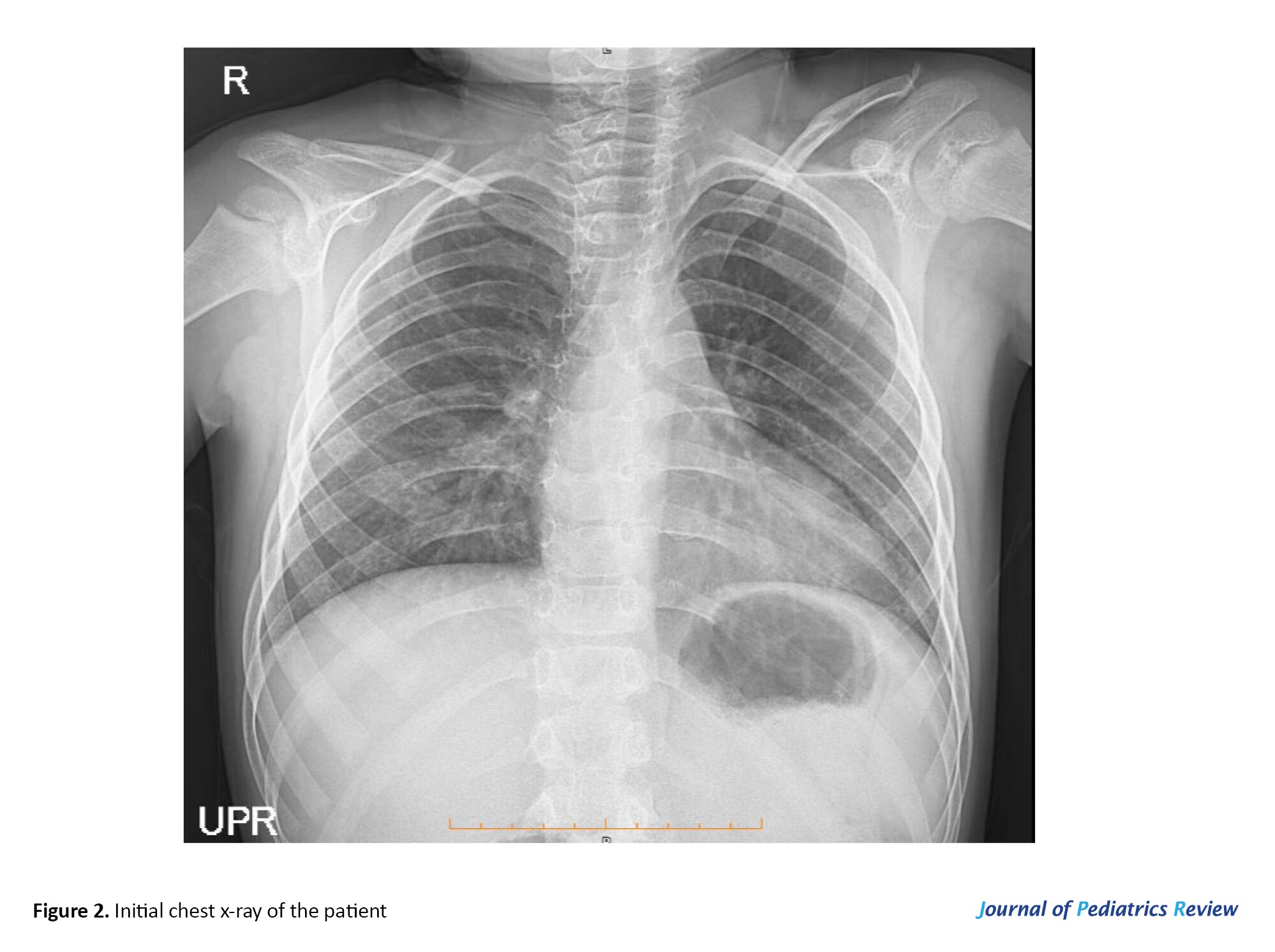
His electrocardiogram was normal, and echocardiography evaluation revealed normal left ventricular systolic function with 60% ejection fraction and normal proximal coronary arteries with no sign of vegetation or clot. However, trivial mitral regurgitation and tricuspid regurgitation were detected. Abdominopelvic sonography was normal, and due to positive P-ANCA and a history of DAH, a paranasal sinus computed tomography scan was ordered, which was intact. Figure 2 presents the initial chest x-ray.
We did a biopsy of the lesions, which declared pauci-inflammatory vacuolar lymphocytic interface dermatitis with pronounced mucinosis in the dermis. Based on the history of arthralgia, low levels of complements, skin lesions, anemia, elevated ESR, and positive anti-dsDNA level, acute SLE was confirmed for him. After consulting with the ophthalmology consultant, treatment with prednisolone and hydroxychloroquine was started. After three months of follow-up, his condition improved, and the lesions healed.
Discussion
The complement system plays an important role in SLE. Its activation by immune complexes drives type III hypersensitivity reactions, causing inflammatory responses in tissues. The complement system also contributes to the clearance of immune complexes and apoptotic cells, which are often elevated in SLE patients. However, the complement system can become dysregulated in SLE, leading to excessive activation and tissue damage, and decreased serum components of C4 and C3 are markers of active disease [2, 5, 6].
JRA encompasses a diverse array of systemic onset diseases with varying clinical manifestations. The condition is categorized based on demographic factors, clinical features, treatment approaches, and disease outlook [7]. A recent study revealed that arthralgias were the most frequently reported initial symptoms in 98.1% of patients, followed by fever (52.1%), fatigue, malaise, morning stiffness (15.5%), and Raynaud phenomenon (0.5%) [8]. In laboratory evaluation, positive RF, ANA, anti-CCP, and inflammatory markers, along with positive HLA-B27, are common findings in patients [7]. The patient in our care presented with symptoms of arthralgia, bone pain, and constitutional symptoms. Additionally, the detection of elevated inflammatory markers, including ESR and CRP, coupled with the absence of other rheumatologic manifestations, initially resulted in a diagnosis of JRA for the patient in his first admission.
Lupus is a persistent and severe autoimmune/inflammatory condition that can impact various organ systems, resulting in significant harm, disability, or even death. When lupus emerges in individuals under the age of 18 years, it is commonly known as childhood-onset SLE (cSLE). This condition is rare, with an estimated incidence of 0.3-0.9 cases per 100000 children annually and a prevalence ranging from 3.3 to 24 cases per 100 000 children. The average age is around 12.6 years, and it predominantly affects females, with a female-to-male ratio of 4:3 in the first decade of life. cSLE typically follows a more severe clinical course compared to adult-onset cases, often presenting with a higher occurrence of complications such as lupus nephritis, hematologic abnormalities, photosensitivity, cardiorespiratory involvements (such as DAH), neuropsychiatric issues, and mucocutaneous involvement [9, 10]. The diagnosis of SLE is based on positive clinical and immunological (such as low complements, positive anti-dsDNA) findings [11]. Our patient was a 3-year-old boy who was later presented with DAH and the presence of low complements (C3, C4) during evaluation. Following a thorough examination of the patient’s rheumatologic and genetic tests, it was determined that the individual did not exhibit the necessary indicators for a diagnosis of SLE. In addition, the patient also had a history of multiple viral and bacterial infections. As a result, the patient was discharged with a diagnosis of hypocomplementemia and prescribed a treatment regimen including prednisolone, calcium supplements, and the rituximab protocol.
Typical laboratory results for children with SLE consist of anemia, thrombocytopenia, decreased levels of vitamin D, and elevated ESR. Autoantibodies like ANA, anti-dsDNA, and anti-phospholipid antibodies are also expected to be present in the serum in these patients. ANA positivity is common in all children with SLE; however, there are some controversial cases of “ANA-negative” SLE. It is crucial to evaluate an ANA-negative child for alternative diagnoses thoroughly. Some SLE patients may lose their positive ANA over time, and seronegativity may be due to technical errors or ANA entrapment in immune complexes. Despite this, ANA-negative patients may still exhibit similar clinical symptoms to their ANA-positive counterparts. Anti-dsDNA and anti-SM autoantibodies can aid in confirming SLE diagnosis. These antibodies can also be found in children who do not meet the SLE classification criteria; some may develop other rheumatic diseases or remain asymptomatic [3, 6, 12].
Patients who develop the condition very early in life are more likely to exhibit an atypical presentation, including a lack of autoantibodies, experience a more severe disease progression, and have a less favorable prognosis. In contrast to adult-onset SLE, cSLE tends to be more aggressive, with increased disease activity and a higher burden of medications, such as corticosteroids and other immunosuppressive drugs, contributing to the elevated morbidity and mortality rates associated with the disease [9, 10, 13]. Our patient developed new scattered maculopapular erythematous and scaling lesions about 6 months ago. During our investigation, rheumatology biomarkers (include, low C3 and C4 levels, elevated P-ANCA level, and positive anti-dsDNA) became positive, report of lesion biopsy, clinical finding (bone pain, skin lesions), and previous history of DAH, arthralgia, and bone pain, met the criteria for the diagnosis of cSLE for the patient. Then patient was treated with prednisolone and hydroxychloroquine, and his skin lesions healed within three months of follow-up.
Conclusion
It is crucial to make an accurate and timely diagnosis because the clinical presentation of SLE patients can range from acute disease, a rapidly fatal illness, to a chronic disorder with an intermittent or continuous course. Patients with SLE commonly present with specific autoantibodies and early disease symptoms, such as constitutional, mucocutaneous, musculoskeletal, hematologic, renal, serositis, and neuropsychiatric symptoms. However, the broad spectrum and unusual clinical manifestations of SLE in susceptible patients should also be considered. Early recognition and intervention are critical to mitigate long-term complications and improve patient outcomes. Furthermore, a multidisciplinary approach involving rheumatologists, pediatricians, and other specialists is often necessary to provide comprehensive care for individuals with SLE.
Study limitations
We encountered limitations in our study. Due to the lack of facilities in our hospital, we had to send biopsy samples and rheumatological tests to another center, potentially causing delays in diagnosis and treatment. Our study’s strengths lie in the rarity of diagnosing this disease in the patient and the scarcity of previous research in this area. With our prompt diagnosis and suitable treatment, the patient’s overall condition has improved without complications.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Research Ethics Committee of Golestan University of Medical Sciences, Gorgan, Iran (Code: IR.GOUMS.REC.1403.211). Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. The purpose of this case report was completely explained to the patient’s legal guardian, and he was assured that the information would be kept confidential by the researchers. This case report was performed in line with principles of the Declaration of Helsinki.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualization: Lobat Shahkar, and Narges Lashkarbolouk; Data collection and writing the original draft: Mahdi Mazandarani, and Narges Lashkarbolouk; Review and editing: All authors.
Conflicts of interest
The authors declared no conflict of interests.
References
Systemic lupus erythematosus (SLE) is a prototype of a multi-system autoimmune disease that arises from a complex etiopathogenesis. Typically, the disease affects the skin, the musculoskeletal system, and the kidneys. The exact cause and mechanism of SLE still need to be fully understood. Still, it is considered to involve a combination of genetic, environmental, and hormonal factors [1, 2].
In children, unusual clinical presentation with uncommon symptoms happens, mainly at a young age (under six years old), and should be considered a chronological history of the family history of autoimmunity and all relevant events. The disease often manifests with signs affecting multiple organ systems in children and adolescents [1, 2]. Early-onset SLE is linked to specific complement deficiencies in the classical pathway (C1, C4, and C2), which may increase susceptibility to SLE at a young age. These deficiencies can contribute to the dysregulation of the immune system, leading to the production of autoantibodies and the development of systemic inflammation characteristic of SLE [3].
This chronic disease is characterized by variability in clinical phenotypes and the production of multiple autoantibodies. The disease presents with a wide range of symptoms and signs, including photosensitivity rash, hair loss, arthritis, arthralgia, hematuria, proteinuria, hypergammaglobulinemia, hypocomplementemia, and positive rheumatological tests. SLE patients may display acute and severe disease with seizure, psychosis, uremia, pancreatitis, pulmonary hemorrhage, or sepsis [3].
The diagnosis of SLE is determined based on a range of clinical symptoms, serological and laboratory tests, biopsies, or other diagnostic procedures. Key diagnostic tools include blood tests for specific antibodies (such as antinuclear antibodies [ANA], anti-double-stranded DNA [anti-dsDNA], and anti-Smith antibody), with elevated levels indicating SLE. Urinalysis can reveal kidney involvement by detecting protein or blood, while imaging techniques like ultrasound, x-rays, or computed tomography scans may assess organ involvement, particularly in the kidneys and lungs [4, 5].
Early diagnosis and treatment are crucial for managing the disease and preventing organ damage. The diagnosis of SLE can be complex, and if SLE is suspected, a comprehensive evaluation and ongoing monitoring are typically needed to confirm the diagnosis and develop an appropriate treatment plan.
Case Presentation
A 7-year-old boy Baloch patient with a past medical history of C3 and C4 hypocomplementemia, was referred to our pediatric hospital with a chief complaint of new skin lesions. He was taking prednisolone, alendronate, and calcium supplements. He had been treated with the rituximab protocol, which was discontinued about six months ago. The lesions have gradually appeared and have intensified over the past month. The lesions were not itchy, and there was no discharge. The patient also reports that the lesions worsened after exposure to light. Moreover, he suffered from arthralgia and bone pain. There was no history of fever, dyspnea, chest pain, or neurological symptoms.
In his physical examination, scattered maculopapular erythematous and scaling lesions were observed in both upper and lower limbs (Figure 1). His vital signs were stable and neurological, and fundus examinations were intact. There was no sign or history of malar rash, arthritis, hepatosplenomegaly, or lymphadenopathy. According to the patient’s parents, due to improvement in their child’s clinical condition and normal complement level, they discontinued rituximab treatment about six months ago.
In the maternal lineage, there were no issues during pregnancy and childbirth, and no birth defects were noted in the individual’s siblings. There was no familial background of genetic susceptibility or hereditary cancer-related conditions. Additionally, his parents do not relate.
In his past medical history, he had a history of bone pain, arthralgia, and intermittent fever when he was 2 years old. Due to the presence of anemia and normal rheumatologic laboratory tests, he was diagnosed with juvenile idiopathic arthritis (JRA) and was treated with methotrexate along with oral corticosteroids. Later, he had an episode of severe dyspnea, low oxygen saturation level, and hemoptysis when he was three years old. He was admitted with an initial diagnosis of diffuse alveolar hemorrhage (DAH). During his hospitalization, there were high levels of C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), anemia, and hypocomplementemia (low C4 and C3). Normal ANA, anti-dsDNA, rheumatoid factor (RF), anti-cyclic citrullinated peptide (anti-CCP), anti-neutrophil cytoplasmic Autoantibody, Cytoplasmic (C-ANCA), perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA), anti-Ro, anti-La, lupus anticoagulant, anti-glomerular basement membrane diseases (Anti-GBM), anti-cardiolipin Ab, anti-phospholipid Ab, and urine analysis were also observed. The diagnosis of hypocomplementemia was confirmed for him. When he was 4 years old, he also had a history of severe pulmonary cytomegalovirus (CMV) infection, which was treated with ganciclovir.
In our evaluation, he had anemia, high ESR level, hematuria, low levels of C3 and C4, elevated P-ANCA level, and positive anti-dsDNA with a normal amount of other rheumatological tests. The viral markers were negative for human immunodeficiency virus (HIV), CMV, hepatitis B virus, and hepatitis C virus (HCV). The immunoglobulin (Ig) M, IgG, IgE, and IgA levels were also in the normal range (Table 1).
His electrocardiogram was normal, and echocardiography evaluation revealed normal left ventricular systolic function with 60% ejection fraction and normal proximal coronary arteries with no sign of vegetation or clot. However, trivial mitral regurgitation and tricuspid regurgitation were detected. Abdominopelvic sonography was normal, and due to positive P-ANCA and a history of DAH, a paranasal sinus computed tomography scan was ordered, which was intact. Figure 2 presents the initial chest x-ray.
We did a biopsy of the lesions, which declared pauci-inflammatory vacuolar lymphocytic interface dermatitis with pronounced mucinosis in the dermis. Based on the history of arthralgia, low levels of complements, skin lesions, anemia, elevated ESR, and positive anti-dsDNA level, acute SLE was confirmed for him. After consulting with the ophthalmology consultant, treatment with prednisolone and hydroxychloroquine was started. After three months of follow-up, his condition improved, and the lesions healed.
Discussion
The complement system plays an important role in SLE. Its activation by immune complexes drives type III hypersensitivity reactions, causing inflammatory responses in tissues. The complement system also contributes to the clearance of immune complexes and apoptotic cells, which are often elevated in SLE patients. However, the complement system can become dysregulated in SLE, leading to excessive activation and tissue damage, and decreased serum components of C4 and C3 are markers of active disease [2, 5, 6].
JRA encompasses a diverse array of systemic onset diseases with varying clinical manifestations. The condition is categorized based on demographic factors, clinical features, treatment approaches, and disease outlook [7]. A recent study revealed that arthralgias were the most frequently reported initial symptoms in 98.1% of patients, followed by fever (52.1%), fatigue, malaise, morning stiffness (15.5%), and Raynaud phenomenon (0.5%) [8]. In laboratory evaluation, positive RF, ANA, anti-CCP, and inflammatory markers, along with positive HLA-B27, are common findings in patients [7]. The patient in our care presented with symptoms of arthralgia, bone pain, and constitutional symptoms. Additionally, the detection of elevated inflammatory markers, including ESR and CRP, coupled with the absence of other rheumatologic manifestations, initially resulted in a diagnosis of JRA for the patient in his first admission.
Lupus is a persistent and severe autoimmune/inflammatory condition that can impact various organ systems, resulting in significant harm, disability, or even death. When lupus emerges in individuals under the age of 18 years, it is commonly known as childhood-onset SLE (cSLE). This condition is rare, with an estimated incidence of 0.3-0.9 cases per 100000 children annually and a prevalence ranging from 3.3 to 24 cases per 100 000 children. The average age is around 12.6 years, and it predominantly affects females, with a female-to-male ratio of 4:3 in the first decade of life. cSLE typically follows a more severe clinical course compared to adult-onset cases, often presenting with a higher occurrence of complications such as lupus nephritis, hematologic abnormalities, photosensitivity, cardiorespiratory involvements (such as DAH), neuropsychiatric issues, and mucocutaneous involvement [9, 10]. The diagnosis of SLE is based on positive clinical and immunological (such as low complements, positive anti-dsDNA) findings [11]. Our patient was a 3-year-old boy who was later presented with DAH and the presence of low complements (C3, C4) during evaluation. Following a thorough examination of the patient’s rheumatologic and genetic tests, it was determined that the individual did not exhibit the necessary indicators for a diagnosis of SLE. In addition, the patient also had a history of multiple viral and bacterial infections. As a result, the patient was discharged with a diagnosis of hypocomplementemia and prescribed a treatment regimen including prednisolone, calcium supplements, and the rituximab protocol.
Typical laboratory results for children with SLE consist of anemia, thrombocytopenia, decreased levels of vitamin D, and elevated ESR. Autoantibodies like ANA, anti-dsDNA, and anti-phospholipid antibodies are also expected to be present in the serum in these patients. ANA positivity is common in all children with SLE; however, there are some controversial cases of “ANA-negative” SLE. It is crucial to evaluate an ANA-negative child for alternative diagnoses thoroughly. Some SLE patients may lose their positive ANA over time, and seronegativity may be due to technical errors or ANA entrapment in immune complexes. Despite this, ANA-negative patients may still exhibit similar clinical symptoms to their ANA-positive counterparts. Anti-dsDNA and anti-SM autoantibodies can aid in confirming SLE diagnosis. These antibodies can also be found in children who do not meet the SLE classification criteria; some may develop other rheumatic diseases or remain asymptomatic [3, 6, 12].
Patients who develop the condition very early in life are more likely to exhibit an atypical presentation, including a lack of autoantibodies, experience a more severe disease progression, and have a less favorable prognosis. In contrast to adult-onset SLE, cSLE tends to be more aggressive, with increased disease activity and a higher burden of medications, such as corticosteroids and other immunosuppressive drugs, contributing to the elevated morbidity and mortality rates associated with the disease [9, 10, 13]. Our patient developed new scattered maculopapular erythematous and scaling lesions about 6 months ago. During our investigation, rheumatology biomarkers (include, low C3 and C4 levels, elevated P-ANCA level, and positive anti-dsDNA) became positive, report of lesion biopsy, clinical finding (bone pain, skin lesions), and previous history of DAH, arthralgia, and bone pain, met the criteria for the diagnosis of cSLE for the patient. Then patient was treated with prednisolone and hydroxychloroquine, and his skin lesions healed within three months of follow-up.
Conclusion
It is crucial to make an accurate and timely diagnosis because the clinical presentation of SLE patients can range from acute disease, a rapidly fatal illness, to a chronic disorder with an intermittent or continuous course. Patients with SLE commonly present with specific autoantibodies and early disease symptoms, such as constitutional, mucocutaneous, musculoskeletal, hematologic, renal, serositis, and neuropsychiatric symptoms. However, the broad spectrum and unusual clinical manifestations of SLE in susceptible patients should also be considered. Early recognition and intervention are critical to mitigate long-term complications and improve patient outcomes. Furthermore, a multidisciplinary approach involving rheumatologists, pediatricians, and other specialists is often necessary to provide comprehensive care for individuals with SLE.
Study limitations
We encountered limitations in our study. Due to the lack of facilities in our hospital, we had to send biopsy samples and rheumatological tests to another center, potentially causing delays in diagnosis and treatment. Our study’s strengths lie in the rarity of diagnosing this disease in the patient and the scarcity of previous research in this area. With our prompt diagnosis and suitable treatment, the patient’s overall condition has improved without complications.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Research Ethics Committee of Golestan University of Medical Sciences, Gorgan, Iran (Code: IR.GOUMS.REC.1403.211). Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. The purpose of this case report was completely explained to the patient’s legal guardian, and he was assured that the information would be kept confidential by the researchers. This case report was performed in line with principles of the Declaration of Helsinki.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualization: Lobat Shahkar, and Narges Lashkarbolouk; Data collection and writing the original draft: Mahdi Mazandarani, and Narges Lashkarbolouk; Review and editing: All authors.
Conflicts of interest
The authors declared no conflict of interests.
References
- Silva CA. Childhood-onset systemic lupus erythematosus: Early disease manifestations that the paediatrician must know. Expert Rev Clin Immunol. 2016; 12(9):907-10. [DOI:10.1080/1744666X.2016.1195685] [PMID]
- Kim AHJ, Strand V, Sen DP, Fu Q, Mathis NL, Schmidt MJ, et al. Association of Blood Concentrations of Complement Split Product iC3b and Serum C3 With Systemic Lupus Erythematosus Disease Activity. Arthritis Rheumatol. 2019; 71(3):420-30. [PMID]
- Raymond W, Eilertsen G, Nossent J. Hypocomplementemia as a risk factor for organ damage accrual in patients with systemic lupus erythematosus. J Immunol Res. 2018; 2018:8051972. [DOI:10.1155/2018/8051972] [PMID]
- Kumar S, Nair S, Rajam L. Case series of pediatric systemic lupus erythematosus from Kerala: Comparison with other Indian series. Int J Rheum Dis. 2010; 13(4):391-5. [DOI:10.1111/j.1756-185X.2010.01536.x] [PMID]
- Aringer M. Inflammatory markers in systemic lupus erythematosus. J Autoimmun. 2020; 110:102374. [DOI:10.1016/j.jaut.2019.102374] [PMID]
- Durcan L, Petri M. The clinical and serological associations of hypocomplementemia in a longitudinal sle cohort. Semin Arthritis Rheum. 2020; 50(5):1081-6. [DOI:10.1016/j.semarthrit.2020.06.009] [PMID]
- Hazazi AS, Alshehri BA, Alali YA, Alqarni RM, Hazazi IS, Albalawi NA, et al. Overview on Juvenile Idiopathic Arthritis: A review. J Pharm Res Int. 2021; 33(40B):139-47. [DOI:10.9734/jpri/2021/v33i40B32273]
- Şen V, Ece A, Uluca Ü, Güneş A, Yel S, Tan I, et al. Evaluation of children with juvenile idiopathic arthritis in southeastern Turkey: A single center experience. Hippokratia. 2015; 19(1):63-8. [PMID]
- Charras A, Smith E, Hedrich CM. Systemic lupus erythematosus in children and young people. Curr Rheumatol Rep. 2021; 23(3):20. [DOI:10.1007/s11926-021-00985-0] [PMID]
- Harry O, Yasin S, Brunner H. Childhood-onset systemic lupus erythematosus: A review and update. J Pediatr. 2018; 196:22-30.e2. [DOI:10.1016/j.jpeds.2018.01.045] [PMID]
- Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019; 71(9):1400-12. [PMID]
- Miguel DF, Terreri MT, Pereira RMR, Bonfá E, Silva CAA, Corrente JE, et al. Comparison of urinary parameters, biomarkers, and outcome of childhood systemic lupus erythematosus early onset-lupus nephritis. Adv Rheumatol. 2020; 60(1):10. [DOI:10.1186/s42358-020-0114-4] [PMID]
- Costagliola G, Mosca M, Migliorini P, Consolini R. Pediatric Systemic Lupus Erythematosus: Learning From Longer Follow Up to Adulthood. Front Pediatr. 2018; 6:144.[DOI:10.3389/fped.2018.00144] [PMID]
Type of Study: Case Report and Review of Literature |
Subject:
Allergy and Clinical Immunology
Received: 2024/07/30 | Accepted: 2025/03/2 | Published: 2025/04/1
Received: 2024/07/30 | Accepted: 2025/03/2 | Published: 2025/04/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
