
Sun, Aug 9, 2026


Volume 14, Issue 1 (1-2026)
J. Pediatr. Rev 2026, 14(1): 81-86 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Sheidaee K, Abbaskhanian A, Mesgarankarimi A. NRROS-associated Microgliopathy With Cerebral Calcification and Resistant Epilepsy: A Case Report. J. Pediatr. Rev 2026; 14 (1) :81-86
URL: http://jpr.mazums.ac.ir/article-1-736-en.html
URL: http://jpr.mazums.ac.ir/article-1-736-en.html



1- Department of Pediatrics, Bu-Ali Sina Hospital, Mazandaran University of Medical Sciences, Sari, Iran. , azadehsheidaee@gmail.com
2- Department of Pediatrics, Bu-Ali Sina Hospital, Mazandaran University of Medical Sciences, Sari, Iran.
2- Department of Pediatrics, Bu-Ali Sina Hospital, Mazandaran University of Medical Sciences, Sari, Iran.
Full-Text [PDF 644 kb]
(300 Downloads)
| Abstract (HTML) (838 Views)
Full-Text: (195 Views)
Background
Negative regulator of reactive oxygen species (NRROS) is a membrane protein that contains leucine-rich repeats in the endoplasmic reticulum (ER) [1]. NRROS is preferentially expressed in myeloid cells, such as macrophages and neutrophils, and regulates reactive oxygen species (ROS) production by controlling NOX2 protein stability. NRROS is highly expressed in CNS-resident macrophages, including microglia and perivascular macrophages (PVMs) [2]. NRROS limits ROS production by phagocytes during inflammatory responses. In addition, ROS levels increase in NRROS-deficient phagocytes in response to inflammatory challenges. NRROS regulate ROS production—a mechanism that enables phagocytes to produce larger amounts of ROS when needed to control invading pathogens while minimizing unwanted collateral tissue damage [3]. Loss of NRROS leads to astrogliosis, impaired motor function, and reduced lifespan. Additionally, during early embryonic development, NRROS expression in microglia is required for differentiation [2].
NRROS was newly recognized as a novel binding partner of latent TGF-β1. Specific binding of NRROS to latent TGF-β1 relieves active TGF-β1 to activate TGF-β signaling in myeloid leukemia cells and THP-1 monocytes. Activation of TGF-β1 is vital in the central nervous system [4, 5]. Increased neuronal cell death and microgliosis have been observed in TGF-β1-deficient mice. The main cause of these abnormalities in NRROS knockout mice is likely decreased TGF-β1 activation in microglia. Hence, these observations suggest that the TGFβ signaling pathway plays a vital role in central nervous system (CNS) development and that abnormal NRROS function can impair neuronal function [6, 7].
NRROS-associated microgliopathy is a recently recognized neurodegenerative condition, characterized by neurodegeneration and drug-resistant epilepsy, and it seems that brain calcification is a specific sign in patients and is the diagnostic key of the disease.
Case Presentation
The patient, an 8-year-old male, is the first and only child of Iranian parents who are first cousins. He was born after a normal pregnancy and delivery. He was a term infant born via elective cesarean delivery with normal perinatal and growth indices and no evident family history of similar symptoms. At birth, the baby weighed 2800 g (between the 3rd and 15th percentiles), was 49 cm long (between the 15th and 50th percentiles), and had a head circumference of 34 cm (between the 15th and 50th percentiles). His early development was unremarkable, with mild global developmental delay beginning, which worsened at 1.5 years of age following the onset of seizures. He gradually lost the ability to walk and talk within a year.
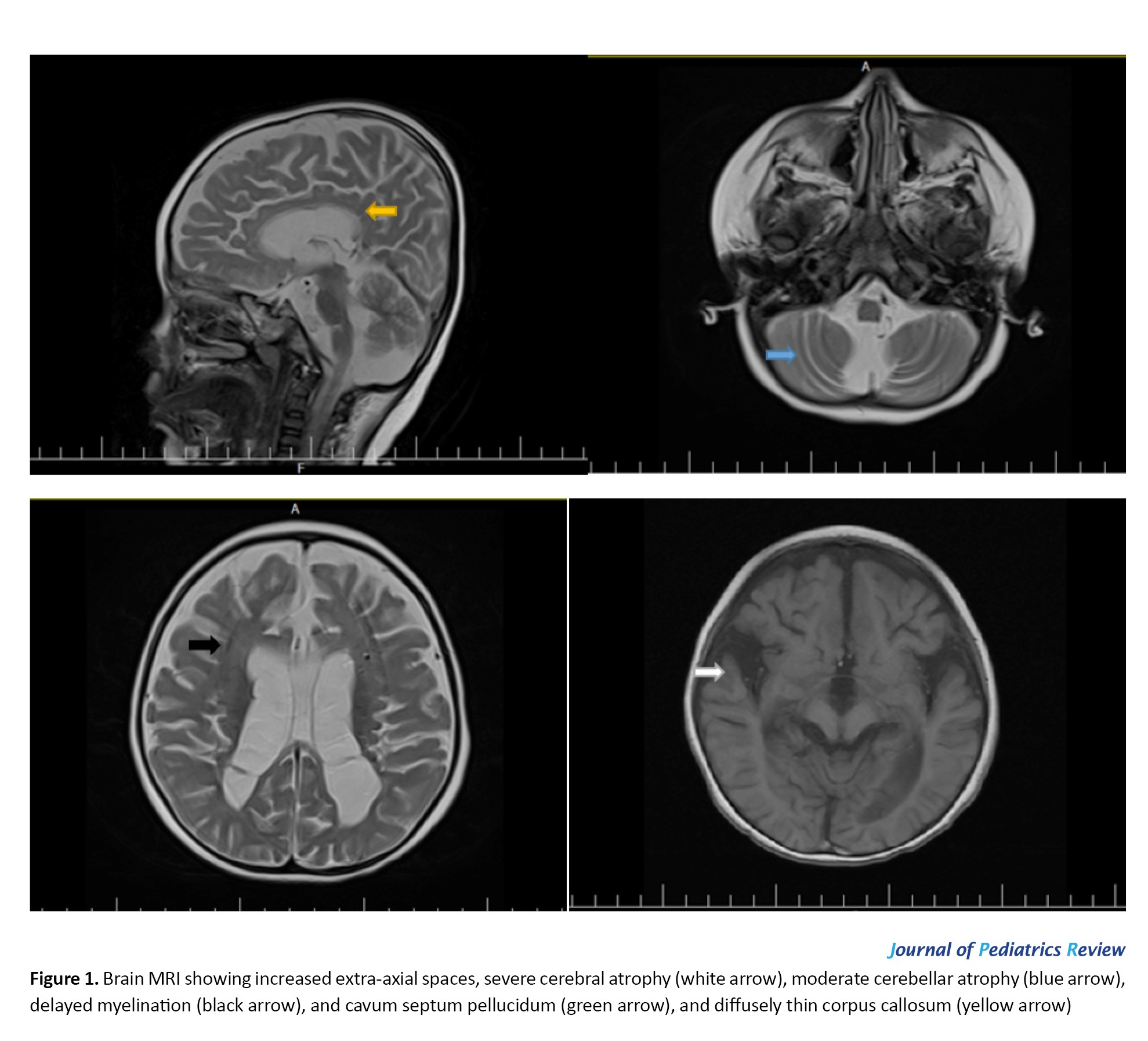
Metabolic testing at the age of 2 years, including ammonia, pyruvate, lactate, serum amino acids, urine organic acids, and acylcarnitine profile, was noncontributory. Brain magnetic resonance imaging (MRI) of the patient at the age of 3 years showed increased extra-axial spaces, severe cerebral and moderate cerebellar atrophy, delayed myelination, and a diffusely thin corpus callosum (Figure 1).
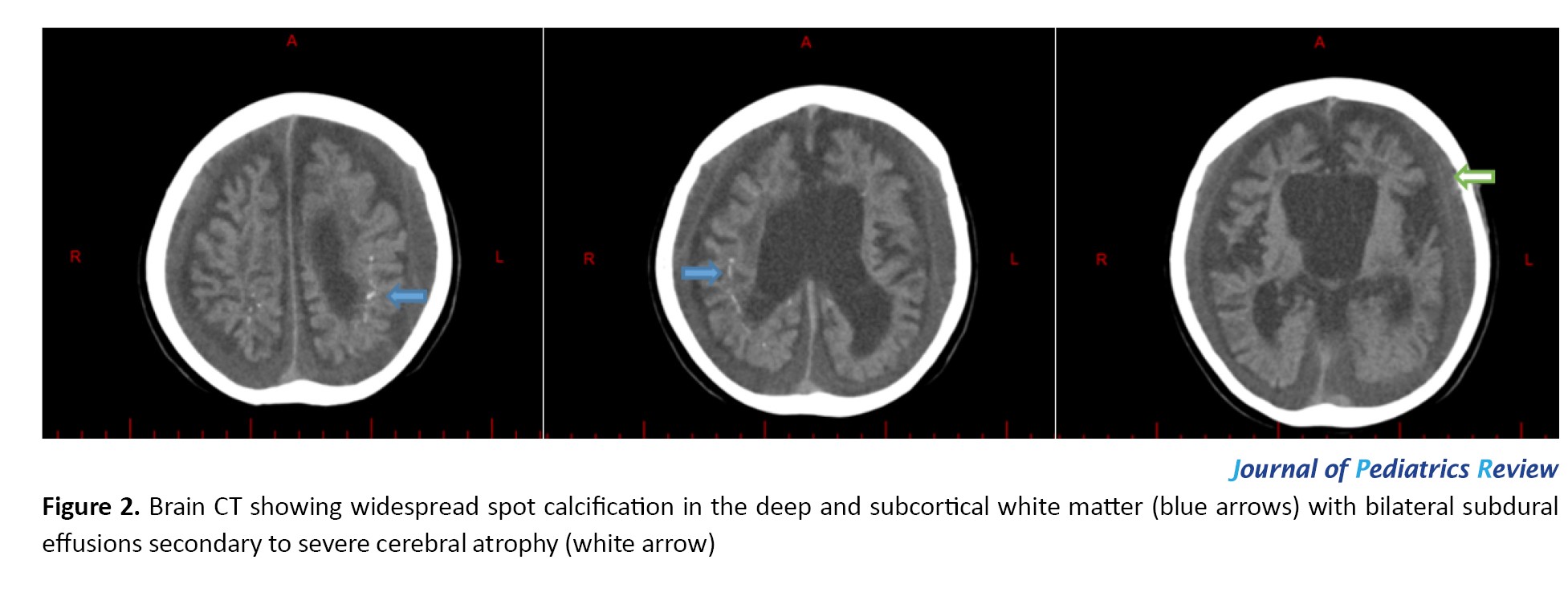
 At first, the seizures were tonic with fever and then mostly myoclonic without any fever, which were all resistant to anticonvulsant drugs. The patient was administered a comprehensive pharmacotherapy regimen, including phenobarbital, levetiracetam, topiramate, sodium valproate, vigabatrin, and clonazepam, each as monotherapy and in combination. Adjunctive therapies, including oral prednisolone, vitamin trials, and intravenous immunoglobulin (IVIG), were also implemented. Despite these extensive therapeutic efforts, the patient did not exhibit significant clinical improvement. Since then, he has had refractory daily seizures and difficulty swallowing and has frequently been hospitalized with these complaints and impressions, such as encephalopathy and aspiration pneumonia. In the last physical examination, the patient was found to have a nondysmorphic face that was spastic, quadriplegic, and axial hypotonic. The patient’s anthropometric measurements revealed a weight of 14 kg, a height of 97 cm, and a head circumference of 48 cm, all falling below the 3rd percentile. The patient was unable to swallow and was fed through a gastrostomy tube. The patient also had a tracheostomy for breathing. Brain computed tomography (CT) images of the patient at the age of 6 years demonstrated widespread spot calcification in the deep and subcortical white matter with bilateral subdural effusions secondary to severe cerebral atrophy (Figure 2).
At first, the seizures were tonic with fever and then mostly myoclonic without any fever, which were all resistant to anticonvulsant drugs. The patient was administered a comprehensive pharmacotherapy regimen, including phenobarbital, levetiracetam, topiramate, sodium valproate, vigabatrin, and clonazepam, each as monotherapy and in combination. Adjunctive therapies, including oral prednisolone, vitamin trials, and intravenous immunoglobulin (IVIG), were also implemented. Despite these extensive therapeutic efforts, the patient did not exhibit significant clinical improvement. Since then, he has had refractory daily seizures and difficulty swallowing and has frequently been hospitalized with these complaints and impressions, such as encephalopathy and aspiration pneumonia. In the last physical examination, the patient was found to have a nondysmorphic face that was spastic, quadriplegic, and axial hypotonic. The patient’s anthropometric measurements revealed a weight of 14 kg, a height of 97 cm, and a head circumference of 48 cm, all falling below the 3rd percentile. The patient was unable to swallow and was fed through a gastrostomy tube. The patient also had a tracheostomy for breathing. Brain computed tomography (CT) images of the patient at the age of 6 years demonstrated widespread spot calcification in the deep and subcortical white matter with bilateral subdural effusions secondary to severe cerebral atrophy (Figure 2).
 According to whole-exome sequencing, the homozygous NRROS variant was reported: NRROS (NM_198565): c.1487delG (p.val498phefs*2), which was proven to be pathogenic according to the ACMG guidelines for the interpretation of sequence variations [8, 9]. These results were confirmed by Sanger DNA sequencing. His parents also underwent Sanger DNA sequencing, which confirmed that they were both carriers of this variant.
According to whole-exome sequencing, the homozygous NRROS variant was reported: NRROS (NM_198565): c.1487delG (p.val498phefs*2), which was proven to be pathogenic according to the ACMG guidelines for the interpretation of sequence variations [8, 9]. These results were confirmed by Sanger DNA sequencing. His parents also underwent Sanger DNA sequencing, which confirmed that they were both carriers of this variant.
Written informed consent was secured from the patient’s parents to publish the case in a medical journal.
Discussion
The findings from this case provide significant insight into the pathophysiology and clinical course of NRROS-associated microgliopathy, a recently identified neurodegenerative condition. NRROS encodes a transmembrane protein essential for regulating reactive oxygen species (ROS) and transforming growth factor beta-1 (TGF-β1) signaling. This case adds to the growing body of evidence that the loss of NRROS function leads to a cascade of neuroinflammatory processes, characterized by uncontrolled microglial activation, which ultimately results in progressive neurodegeneration, drug-resistant epilepsy, and cerebral calcifications [10, 11].
The hallmark of intracranial calcifications observed on brain imaging emerges as a critical diagnostic feature of NRROS-associated disorders. In this patient, widespread calcifications in the deep and subcortical white matter, along with cerebral atrophy, were crucial in guiding the diagnosis. This finding is consistent with previous reports, where calcifications serve as an early radiological indicator, often appearing before the full manifestation of neurodegenerative symptoms. Recognizing this calcification pattern in early neuroimaging can expedite diagnosis and prevent further invasive investigations, allowing clinicians to focus on tailored therapeutic strategies sooner.
Twelve individuals with biallelic NRROS variants have been reported to date, as summarized below. The following has been explained about these patients (Table 1).
Smith et al. [12] ascertained 3 patients demonstrating a stereotyped clinical and neuroradiological phenotype. All three children were born after normal pregnancies and deliveries, and early development was unremarkable. However, in the second year of life, patients experienced refractory seizures and neurodegeneration, leading to death between the ages of 27 and 36 months. Neuroimaging initially demonstrated fine calcification at the depths of the cerebral gyri, with normal white matter. As the disease progresses, repeat imaging reveals increased calcification, severe generalized atrophy with ventricular dilatation, and diffuse signal changes in cerebral and cerebellar white matter [12].
Dong et al. [13] reported 6 patients from four families with biallelic germline variants in the NRROS gene. Development in infancy alternated from normal to moderate global developmental delay. All patients were hypotonic early in life, and five later developed hypertonia that was mainly peripheral. In all the patients, seizures started before the age of one year, and the first seizure type included febrile seizures, focal seizures, infantile spasms, and myoclonic seizures. All the patients had severe and progressive developmental regression following seizure onset. All patients were nondysmorphic. Brain MRI showed obvious cerebral atrophy and delayed myelination in all patients, and corpus callosum hypoplasia was observed in half of the patients. In all patients who had cranial CT scans (3 out of 6), that imaging modality identified multiple sparse, punctate calcifications in the cerebral white matter. Three people passed away when they were between two and four years old. The eldest remaining child was nine years old [13].
Madaan et al. reported that a 2-year-old boy presented with seizures and developmental regression beginning at the age of 9 months. He was developmentally normal until 9 months of age, when he started having seizures after a brief febrile illness. Seizures were occasional initially but were associated with developmental regression. There was an associated progressive loss of all acquired skills, hearing, and vision. CT of the brain (done on day 2 of life) confirmed that colpocephaly was noticed initially via in utero ultrasound and also revealed periventricular and subcortical calcifications [14].
Macintosh et al. reported one patient with a severe neurodegenerative phenotype in which exome sequencing recognized 2 novel variants in NRROS, a missense variant (c.185T>C, p.Leu62Pro) and a premature stop codon (c.310C>T, p.Gln104Ter). Pathological examination revealed extensive involvement of gray and white matter, dystrophic calcifications, and infiltration of foamy macrophages. This case was the first reported patient with NRROS variants and a mitochondrial ultrastructural abnormality, as demonstrated by electron microscopy of autopsy tissue [15].
Kapat et al. evaluated a child with various types of seizures, initially containing complex febrile seizures, followed by afebrile seizures. He had truncal hypotonia, but his appendicular hypotonia progressed to hypertonia over the next few months and further to decorticate posturing. Brain MRI revealed generalized atrophy, and CT showed speckled calcification in the subcortical white matter. His cerebrospinal fluid had normal cytology and biochemical results but was positive for anti-gamma-aminobutyric acid B antibodies. Whole-exome sequencing revealed a likely pathogenic, novel autosomal recessive homozygous variation in the NRROS gene on chromosome 3 [c.1487G>A (p.Trp496Ter)], resulting in a proinflammatory state within the central nervous system and thus promoting autoimmune encephalitis. He was treated with both pulse methylprednisolone and IVIG, followed by a slow taper of oral prednisolone and monthly IVIG infusions. This case was the first report of autoimmune encephalitis triggered by this variation in a child [16].
Our case, involving a homozygous NRROS mutation, further solidifies the connection between biallelic NRROS variants and the severe, progressive neurodegenerative phenotype. The patient’s early development was mildly delayed, but rapid deterioration followed the onset of seizures at 1.5 years of age. The refractory nature of the seizures, combined with developmental regression, highlights the devastating progression typical of this disorder. Despite extensive pharmacotherapy, the patient’s seizures remained unmanageable, emphasizing the limitations of current treatment options for NRROS-related epilepsy.
The absence of effective treatments underscores the importance of early detection. Given that intracranial calcifications appear early in the disease course, clinicians should prioritize brain imaging in infants and young children presenting with global developmental delay, hypotonia, or early-onset seizures. Early diagnosis may allow for closer monitoring of disease progression and the timely initiation of supportive therapies, although no current therapies directly target the underlying microgliopathy caused by NRROS mutations. As research advances, there is hope that early identification of affected individuals could allow for future interventions that may modify the disease course, potentially delaying neurodegeneration and improving quality of life.
This case also contributes novel insights into the genotype-phenotype correlation of NRROS-associated disorders. The patient’s specific mutation, c.1487delG (p.Val498Phefs*2), is distinct from other reported variants, suggesting a possible mutation-specific impact on disease severity and progression. As more cases are reported, it will become clearer whether different NRROS mutations correlate with variable phenotypic severity or disease trajectories. These insights will be critical for genetic counseling and the development of mutation-specific therapeutic approaches.
Conclusion
In conclusion, this case highlights the recognition of NRROS-associated microgliopathy as a distinct clinical entity, with intracranial calcifications as a pivotal diagnostic hallmark. Early neuroimaging plays a crucial role in identifying these patients, and although treatment options remain limited, early detection is essential for monitoring and managing disease progression. This case not only broadens the clinical spectrum of NRROS-associated neurodegeneration but also highlights the need for continued research into targeted therapies that address the underlying pathophysiological mechanisms of this devastating disorder.
Ethical Considerations
Compliance with ethical guidelines
Need for institutional review boards approval was waived for this retrospective case report.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Review and editing: Kobra Sheidaee and Ali Abbaskhanian; Writing the original draft: Amirhosein Mesgarankarimi.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors wish to thank the patient and his family for their participation in this study.
References
Negative regulator of reactive oxygen species (NRROS) is a membrane protein that contains leucine-rich repeats in the endoplasmic reticulum (ER) [1]. NRROS is preferentially expressed in myeloid cells, such as macrophages and neutrophils, and regulates reactive oxygen species (ROS) production by controlling NOX2 protein stability. NRROS is highly expressed in CNS-resident macrophages, including microglia and perivascular macrophages (PVMs) [2]. NRROS limits ROS production by phagocytes during inflammatory responses. In addition, ROS levels increase in NRROS-deficient phagocytes in response to inflammatory challenges. NRROS regulate ROS production—a mechanism that enables phagocytes to produce larger amounts of ROS when needed to control invading pathogens while minimizing unwanted collateral tissue damage [3]. Loss of NRROS leads to astrogliosis, impaired motor function, and reduced lifespan. Additionally, during early embryonic development, NRROS expression in microglia is required for differentiation [2].
NRROS was newly recognized as a novel binding partner of latent TGF-β1. Specific binding of NRROS to latent TGF-β1 relieves active TGF-β1 to activate TGF-β signaling in myeloid leukemia cells and THP-1 monocytes. Activation of TGF-β1 is vital in the central nervous system [4, 5]. Increased neuronal cell death and microgliosis have been observed in TGF-β1-deficient mice. The main cause of these abnormalities in NRROS knockout mice is likely decreased TGF-β1 activation in microglia. Hence, these observations suggest that the TGFβ signaling pathway plays a vital role in central nervous system (CNS) development and that abnormal NRROS function can impair neuronal function [6, 7].
NRROS-associated microgliopathy is a recently recognized neurodegenerative condition, characterized by neurodegeneration and drug-resistant epilepsy, and it seems that brain calcification is a specific sign in patients and is the diagnostic key of the disease.
Case Presentation
The patient, an 8-year-old male, is the first and only child of Iranian parents who are first cousins. He was born after a normal pregnancy and delivery. He was a term infant born via elective cesarean delivery with normal perinatal and growth indices and no evident family history of similar symptoms. At birth, the baby weighed 2800 g (between the 3rd and 15th percentiles), was 49 cm long (between the 15th and 50th percentiles), and had a head circumference of 34 cm (between the 15th and 50th percentiles). His early development was unremarkable, with mild global developmental delay beginning, which worsened at 1.5 years of age following the onset of seizures. He gradually lost the ability to walk and talk within a year.
Metabolic testing at the age of 2 years, including ammonia, pyruvate, lactate, serum amino acids, urine organic acids, and acylcarnitine profile, was noncontributory. Brain magnetic resonance imaging (MRI) of the patient at the age of 3 years showed increased extra-axial spaces, severe cerebral and moderate cerebellar atrophy, delayed myelination, and a diffusely thin corpus callosum (Figure 1).
Written informed consent was secured from the patient’s parents to publish the case in a medical journal.
Discussion
The findings from this case provide significant insight into the pathophysiology and clinical course of NRROS-associated microgliopathy, a recently identified neurodegenerative condition. NRROS encodes a transmembrane protein essential for regulating reactive oxygen species (ROS) and transforming growth factor beta-1 (TGF-β1) signaling. This case adds to the growing body of evidence that the loss of NRROS function leads to a cascade of neuroinflammatory processes, characterized by uncontrolled microglial activation, which ultimately results in progressive neurodegeneration, drug-resistant epilepsy, and cerebral calcifications [10, 11].
The hallmark of intracranial calcifications observed on brain imaging emerges as a critical diagnostic feature of NRROS-associated disorders. In this patient, widespread calcifications in the deep and subcortical white matter, along with cerebral atrophy, were crucial in guiding the diagnosis. This finding is consistent with previous reports, where calcifications serve as an early radiological indicator, often appearing before the full manifestation of neurodegenerative symptoms. Recognizing this calcification pattern in early neuroimaging can expedite diagnosis and prevent further invasive investigations, allowing clinicians to focus on tailored therapeutic strategies sooner.
Twelve individuals with biallelic NRROS variants have been reported to date, as summarized below. The following has been explained about these patients (Table 1).
Smith et al. [12] ascertained 3 patients demonstrating a stereotyped clinical and neuroradiological phenotype. All three children were born after normal pregnancies and deliveries, and early development was unremarkable. However, in the second year of life, patients experienced refractory seizures and neurodegeneration, leading to death between the ages of 27 and 36 months. Neuroimaging initially demonstrated fine calcification at the depths of the cerebral gyri, with normal white matter. As the disease progresses, repeat imaging reveals increased calcification, severe generalized atrophy with ventricular dilatation, and diffuse signal changes in cerebral and cerebellar white matter [12].
Dong et al. [13] reported 6 patients from four families with biallelic germline variants in the NRROS gene. Development in infancy alternated from normal to moderate global developmental delay. All patients were hypotonic early in life, and five later developed hypertonia that was mainly peripheral. In all the patients, seizures started before the age of one year, and the first seizure type included febrile seizures, focal seizures, infantile spasms, and myoclonic seizures. All the patients had severe and progressive developmental regression following seizure onset. All patients were nondysmorphic. Brain MRI showed obvious cerebral atrophy and delayed myelination in all patients, and corpus callosum hypoplasia was observed in half of the patients. In all patients who had cranial CT scans (3 out of 6), that imaging modality identified multiple sparse, punctate calcifications in the cerebral white matter. Three people passed away when they were between two and four years old. The eldest remaining child was nine years old [13].
Madaan et al. reported that a 2-year-old boy presented with seizures and developmental regression beginning at the age of 9 months. He was developmentally normal until 9 months of age, when he started having seizures after a brief febrile illness. Seizures were occasional initially but were associated with developmental regression. There was an associated progressive loss of all acquired skills, hearing, and vision. CT of the brain (done on day 2 of life) confirmed that colpocephaly was noticed initially via in utero ultrasound and also revealed periventricular and subcortical calcifications [14].
Macintosh et al. reported one patient with a severe neurodegenerative phenotype in which exome sequencing recognized 2 novel variants in NRROS, a missense variant (c.185T>C, p.Leu62Pro) and a premature stop codon (c.310C>T, p.Gln104Ter). Pathological examination revealed extensive involvement of gray and white matter, dystrophic calcifications, and infiltration of foamy macrophages. This case was the first reported patient with NRROS variants and a mitochondrial ultrastructural abnormality, as demonstrated by electron microscopy of autopsy tissue [15].
Kapat et al. evaluated a child with various types of seizures, initially containing complex febrile seizures, followed by afebrile seizures. He had truncal hypotonia, but his appendicular hypotonia progressed to hypertonia over the next few months and further to decorticate posturing. Brain MRI revealed generalized atrophy, and CT showed speckled calcification in the subcortical white matter. His cerebrospinal fluid had normal cytology and biochemical results but was positive for anti-gamma-aminobutyric acid B antibodies. Whole-exome sequencing revealed a likely pathogenic, novel autosomal recessive homozygous variation in the NRROS gene on chromosome 3 [c.1487G>A (p.Trp496Ter)], resulting in a proinflammatory state within the central nervous system and thus promoting autoimmune encephalitis. He was treated with both pulse methylprednisolone and IVIG, followed by a slow taper of oral prednisolone and monthly IVIG infusions. This case was the first report of autoimmune encephalitis triggered by this variation in a child [16].
Our case, involving a homozygous NRROS mutation, further solidifies the connection between biallelic NRROS variants and the severe, progressive neurodegenerative phenotype. The patient’s early development was mildly delayed, but rapid deterioration followed the onset of seizures at 1.5 years of age. The refractory nature of the seizures, combined with developmental regression, highlights the devastating progression typical of this disorder. Despite extensive pharmacotherapy, the patient’s seizures remained unmanageable, emphasizing the limitations of current treatment options for NRROS-related epilepsy.
The absence of effective treatments underscores the importance of early detection. Given that intracranial calcifications appear early in the disease course, clinicians should prioritize brain imaging in infants and young children presenting with global developmental delay, hypotonia, or early-onset seizures. Early diagnosis may allow for closer monitoring of disease progression and the timely initiation of supportive therapies, although no current therapies directly target the underlying microgliopathy caused by NRROS mutations. As research advances, there is hope that early identification of affected individuals could allow for future interventions that may modify the disease course, potentially delaying neurodegeneration and improving quality of life.
This case also contributes novel insights into the genotype-phenotype correlation of NRROS-associated disorders. The patient’s specific mutation, c.1487delG (p.Val498Phefs*2), is distinct from other reported variants, suggesting a possible mutation-specific impact on disease severity and progression. As more cases are reported, it will become clearer whether different NRROS mutations correlate with variable phenotypic severity or disease trajectories. These insights will be critical for genetic counseling and the development of mutation-specific therapeutic approaches.
Conclusion
In conclusion, this case highlights the recognition of NRROS-associated microgliopathy as a distinct clinical entity, with intracranial calcifications as a pivotal diagnostic hallmark. Early neuroimaging plays a crucial role in identifying these patients, and although treatment options remain limited, early detection is essential for monitoring and managing disease progression. This case not only broadens the clinical spectrum of NRROS-associated neurodegeneration but also highlights the need for continued research into targeted therapies that address the underlying pathophysiological mechanisms of this devastating disorder.
Ethical Considerations
Compliance with ethical guidelines
Need for institutional review boards approval was waived for this retrospective case report.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Review and editing: Kobra Sheidaee and Ali Abbaskhanian; Writing the original draft: Amirhosein Mesgarankarimi.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors wish to thank the patient and his family for their participation in this study.
References
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. 2016; 17(12):1397-406. [DOI:10.1038/ni.3585] [PMID]
- Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol. 2017; 18(6):633-41. [DOI:10.1038/ni.3743] [PMID]
- Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. 2014; 509(7499):235-9. [DOI:10.1038/nature13152] [PMID]
- Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A milieu molecule for TGF-β required for microglia function in the nervous system. Cell. 2018; 174(1):156-71. e16. [DOI:10.1016/j.cell.2018.05.027] [PMID]
- Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017; 23(9):1018-27. [DOI:10.1038/nm.4397] [PMID]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-β1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003; 40(6):1133-45. [DOI:10.1016/S0896-6273(03)00766-9] [PMID]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014; 17(1):131-43. [DOI:10.1038/nn.3599] [PMID]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17(5):405-24. [DOI:10.1038/gim.2015.30] [PMID]
- Masson E, Zou WB, Génin E, Cooper DN, Le Gac G, Fichou Y, et al. Expanding ACMG variant classification guidelines into a general framework. Hum Genomics. 2022; 16(1):31.[DOI:10.1186/s40246-022-00407-x] [PMID]
- Ng A, Xavier RJ. Leucine-rich repeat (LRR) proteins: Integrators of pattern recognition and signaling in immunity. Autophagy. 2011; 7(9):1082-4. [DOI:10.4161/auto.7.9.16464] [PMID]
- Kim JH, Kim K, Kim I, Seong S, Kim N. NRROS negatively regulates osteoclast differentiation by inhibiting RANKL-mediated NF-κB and reactive oxygen species pathways. Mol Cells. 2015; 38(10):904-10. [DOI:10.14348/molcells.2015.0177] [PMID]
- Smith C, McColl BW, Patir A, Barrington J, Armishaw J, Clarke A, et al. Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol. 2020; 139(5):947-51. [DOI:10.1007/s00401-020-02137-7] [PMID]
- Dong X, Tan NB, Howell KB, Barresi S, Freeman JL, Vecchio D, et al. Bi-allelic LoF NRROS variants impairing active TGF-β1 delivery cause a severe infantile-onset neurodegenerative condition with intracranial calcification. Am J Hum Genet. 2020; 106(4):559-69. [DOI:10.1016/j.ajhg.2020.02.014] [PMID]
- Madaan P, Kaushal Y, Srivastava P, Crow YJ, Livingston JH, Ahuja C, et al. Delineating the epilepsy phenotype of NRROS-related microgliopathy: A case report and literature review. Seizure. 2022; 100:15-20. [DOI:10.1016/j.seizure.2022.06.001] [PMID]
- Macintosh J, Derksen A, Poulin C, Braverman N, Vanderver A, Thiffault I, et al. Novel biallelic variants in NRROS associated with a lethal microgliopathy, brain calcifications, and neurodegeneration. Neurogenetics. 2022; 23(2):151-6. [DOI:10.1007/s10048-022-00683-8] [PMID]
- Kapat A, Pandit A, Das S, Paul DK, Mandal AK, Bala AK. Anti-Gamma aminobutyric acid B autoimmune encephalitis in an Indian child with early-onset seizures, neurodegeneration, and brain calcification due to NRROS Variation: The first reported case worldwide. J Pediatr Epilepsy. 2023; 12(03):109-14. [DOI:10.1055/s-0042-1758147]
Type of Study: Case Report and Review of Literature |
Subject:
Pediatric Neurology
Received: 2025/03/2 | Accepted: 2025/07/23 | Published: 2026/01/1
Received: 2025/03/2 | Accepted: 2025/07/23 | Published: 2026/01/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
