
Mon, Jul 13, 2026


Volume 14, Issue 2 (April 2026)
J. Pediatr. Rev 2026, 14(2): 111-144 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Alian F, Bagheri N, Fayyazpour P, Torki Z, Hashemipour M, Salehi M, et al . Frequency of G6PC1 and SLC37A4 Genetic Variants in Asian Patients With Glycogen Storage Disease Type I: A Systematic Review. J. Pediatr. Rev 2026; 14 (2) :111-144
URL: http://jpr.mazums.ac.ir/article-1-786-en.html
URL: http://jpr.mazums.ac.ir/article-1-786-en.html
Fatemeh Alian1 

 , Nastaran Bagheri2 , Parisa Fayyazpour3 , Zahra Torki4 , Mahin Hashemipour5 , Mansoor Salehi2 , Saeideh Abdolahpour6 , Tayebeh Ranjbarnejad7 , Silva Hovsepian *8 , Noushin Rostampour5
, Nastaran Bagheri2 , Parisa Fayyazpour3 , Zahra Torki4 , Mahin Hashemipour5 , Mansoor Salehi2 , Saeideh Abdolahpour6 , Tayebeh Ranjbarnejad7 , Silva Hovsepian *8 , Noushin Rostampour5
, Nastaran Bagheri2 , Parisa Fayyazpour3 , Zahra Torki4 , Mahin Hashemipour5 , Mansoor Salehi2 , Saeideh Abdolahpour6 , Tayebeh Ranjbarnejad7 , Silva Hovsepian *8 , Noushin Rostampour5
1- Department of Biochemistry, Institute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran. & Isfahan Endocrine and Metabolism Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
2- Cellular, Molecular and Genetics Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
3- Department of Clinical Biochemistry, Faculty of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran. & Student Research Committee, Hamadan University of Medical Sciences, Hamadan, Iran.
4- Department of Medical Genetics, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran.
5- Isfahan Endocrine and Metabolism Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
6- Growth and Development Research Center, Tehran University of Medical Sciences, Tehran, Iran.
7- Medical Biology Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran.
8- Growth and Development Research Center, Tehran University of Medical Sciences, Tehran, Iran. ,silvahovsepsecret@gmail.com
2- Cellular, Molecular and Genetics Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
3- Department of Clinical Biochemistry, Faculty of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran. & Student Research Committee, Hamadan University of Medical Sciences, Hamadan, Iran.
4- Department of Medical Genetics, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran.
5- Isfahan Endocrine and Metabolism Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
6- Growth and Development Research Center, Tehran University of Medical Sciences, Tehran, Iran.
7- Medical Biology Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran.
8- Growth and Development Research Center, Tehran University of Medical Sciences, Tehran, Iran. ,
Keywords: Glycogen storage disease type I (GSD I), Von Gierke disease, G6PC1, SLC37A4, Genetic variation, Asian populations, Systematic review
Full-Text [PDF 986 kb]
(109 Downloads)
| Abstract (HTML) (294 Views)
Full-Text: (85 Views)
Introduction
Glycogen storage diseases are a group of rare metabolic disorders characterized by defects in glycogen metabolism, leading to glycogen accumulation in tissues, particularly the liver and muscles [1-3]. Among more than 20 subtypes, glycogen storage disease type I (GSD I), or von Gierke disease, is the most common [4]. It results from deficiencies in the glucose-6-phosphatase complex (G6PC1), which plays a critical role in the final step of both glycogenolysis and gluconeogenesis by catalyzing the conversion of glucose-6-phosphate to free glucose. This reaction is essential for maintaining normal blood glucose levels during fasting. Deficiency of this complex prevents the release of glucose into the circulation, leading to severe fasting hypoglycemia and intracellular accumulation of glycogen and lipids, particularly in hepatocytes and renal tubular cells. Based on the affected component of the G6Pase complex, GSD I is classified into two major subtypes: GSD Ia, due to G6PC1 deficiency, and GSD Ib, caused by glucose-6-phosphate transporter (SLC37A4) deficiency. GSD I follows an autosomal recessive inheritance pattern and is more prevalent in populations with high rates of consanguinity [3]. Clinically, GSD I presents in infancy with hypoglycemia, lactic acidemia, hyperlipidemia, hyperuricemia, and hepatomegaly. Chronic metabolic imbalance may lead to long-term complications, including growth retardation, hepatic adenomas with a potential risk of malignant transformation, progressive renal dysfunction, osteoporosis, and delayed puberty. GSD Ib also features neutropenia and inflammatory bowel disease, which predispose affected individuals to recurrent bacterial infections, oral ulcers, and inflammatory bowel disease, thereby contributing to increased morbidity and reduced quality of life [1, 3, 5-7].
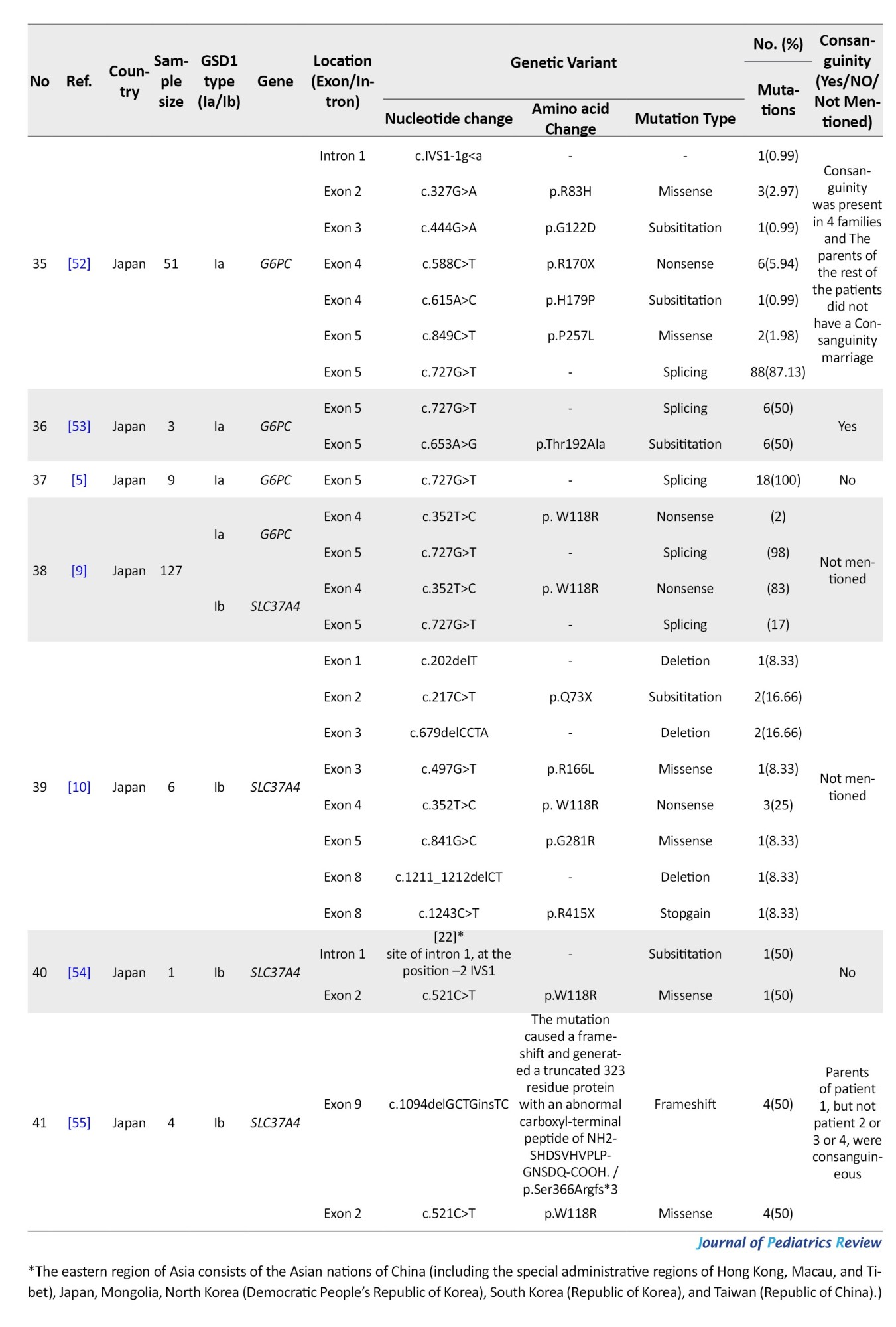
Accurate and early diagnosis is essential to prevent complications. While clinical features and biochemical tests are informative, overlapping symptoms with other GSD types, such as GSD III, can complicate diagnosis. Thus, molecular genetic testing, particularly gene mutation analysis, provides a reliable, noninvasive diagnostic tool [5, 8]. Numerous mutations in G6PC1 and SLC37A4 have been reported across various ethnicities. However, the mutation spectrum and frequencies can differ significantly by region [3]. Despite this genetic diversity, the overall incidence of GSD I does not appear to differ significantly among populations [3].
Although numerous mutations of G6PC1 and SLC37A4 have been reported in various ethnic groups, there is limited systematic data on their prevalence and regional patterns in Asian populations, which hinders optimal genetic counseling and the development of effective diagnostic strategies. This study aims to systematically review reported variants of G6PC1 and SLC37A4 in Asian patients with GSD I. Specifically, we sought to identify prevalent mutations in different Asian regions (participants), compare mutation distributions across regions (comparisons), summarize variant frequencies and patterns (outcomes), and synthesize data from observational studies including case reports, case series, and cohort studies (study design). Identifying prevalent mutations and regional patterns can enhance genetic counseling and streamline diagnostic approaches. These findings may also improve patient outcomes through targeted treatment strategies.
Materials and Methods
Information sources and search strategy
This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and meta-analyses (PRISMA) guidelines. A detailed review protocol was developed a priori and prospectively registered in the international prospective register of systematic reviews (PROSPERO) under the registration number CRD42023475081. The registered protocol is publicly available in the PROSPERO database [1]. A comprehensive literature search was conducted in the following electronic databases: Web of Science, PubMed/MEDLINE, Embase, Scopus, and ProQuest, covering studies published from January 1995 to June 4, 2023. The final search was performed on June 4, 2023. In addition, Google Scholar was searched to identify potentially relevant gray literature.
Reference lists of all included articles and relevant reviews were manually screened to identify additional eligible studies. No direct contact with study authors was undertaken to obtain unpublished data. The full electronic search strategies for all databases, including search terms, Boolean operators, and applied limits (language), are provided in Supplementary File S1 to ensure search reproducibility.
Eligibility criteria
Eligible studies were selected based on the PICOS framework.
Participants (P): Asian patients with a confirmed diagnosis of GSD Ia or GSD Ib.
Intervention/Exposure (I): Genetic analysis of G6PC1 and/or SLC37A4 variants.
Comparison (C): Comparison of variant distribution across different Asian regions and populations, where applicable.
Outcomes (O): Type, frequency, and distribution of pathogenic or likely pathogenic variants.
Study design (S): Observational studies, including case reports, case series, cross-sectional, case–control, and cohort studies.
Studies published between 1995 and 2023 in English and available as full-text peer-reviewed articles were included. Conference abstracts, reviews, editorials, non-English publications, and duplicate reports were excluded to ensure data completeness and methodological quality.
Study selection
All records identified through database searching were imported into reference management software, and duplicate records were removed. Two reviewers independently screened the titles and abstracts of all retrieved studies to identify potentially eligible articles. Full texts of the selected studies were then assessed for eligibility based on the predefined inclusion and exclusion criteria. Any disagreements between reviewers at any stage were resolved through discussion or consultation with a third reviewer. Studies meeting the eligibility criteria were included in the final systematic review. As no meta-analysis was conducted, all eligible studies were synthesized narratively.
Data collection process
Data were independently extracted by two reviewers using a predefined data extraction form. Extracted information included publication year, country, sample size, disease subtype, gene variant details, and consanguinity. Any discrepancies between reviewers were resolved through discussion or consultation with a third reviewer. For studies with missing or unclear information, attempts were made to clarify data from the original reports. This process ensured consistency, accuracy, and reliability of the extracted data.
Data items
The following variables were extracted from each included study:
• Study characteristics: authors, publication year, and country;
• Participants: number of patients, disease subtype (GSD Ia or Ib), consanguinity;
• Gene variants: gene name, nucleotide and protein changes, exon location, type of mutation (missense, nonsense, frameshift, splice-site, deletion).
PICOS elements:
• Participants: Asian patients with GSD I;
• Intervention: molecular genetic testing (gene mutation analysis);
• Comparison: variant distribution across regions;
• Outcome: variant frequencies and patterns;
• Study design: observational studies including case reports, case series, and cohort studies;
Only pathogenic or likely pathogenic variants were included; benign variants and polymorphisms were excluded. No additional assumptions or simplifications were applied beyond the predefined criteria.
Data extraction and regional classification
Two reviewers independently screened and extracted data, with discrepancies resolved by a third reviewer. Extracted data included publication year, country, number of patients, disease subtype, mutation details, and consanguinity. To assess geographic variation, studies were grouped into three Asian regions: East Asia, South Asia, and West Asia, following standard United Nations regional definitions.
Only pathogenic or likely pathogenic mutations were included; benign variants and polymorphisms were excluded from the analysis. Pathogenicity classification was performed based on the American College of Medical Genetics and Genomics (ACMG) guidelines, with ClinVar and VarSome used as reference tools to support and validate variant interpretation. In this systematic review, only variants classified as pathogenic or likely pathogenic were included, while benign variants and polymorphisms were excluded.
Risk of bias within studies
Given the descriptive and genetic nature of the included studies—most of which consisted of case reports, case series, cross-sectional studies, and observational cohorts without comparative interventions or outcome measures—a formal quantitative risk-of-bias assessment was considered of limited applicability. Therefore, study quality was evaluated qualitatively at the study level by assessing the clarity of GSD I diagnosis, confirmation of molecular genetic testing, completeness of variant reporting, and transparency of methodological descriptions. Only studies reporting pathogenic or likely pathogenic variants were included to enhance data reliability. These quality considerations were considered during the narrative synthesis and interpretation of variant distribution and frequency patterns.
Risk of bias across studies
As no meta-analysis was performed, statistical assessments of bias across studies, such as publication bias or heterogeneity measures, were not applicable. Nevertheless, potential sources of bias across studies were considered qualitatively, including uneven geographic representation of Asian countries, small sample sizes in some regions, and selective reporting of variants in individual studies. These limitations are acknowledged and discussed in the Discussion section to provide context for interpreting the overall findings.
Data synthesis and summary measures
As this systematic review did not perform a meta-analysis, results were synthesized narratively and using descriptive statistics. Data from individual studies were extracted and grouped by gene (G6PC1 or SLC37A4), country, and Asian region. Principal summary measures included the number of patients with each variant, allele frequencies, and percentage ranges. Frequencies were presented in tables and summarized narratively to compare variant distributions across Asian regions, highlighting prevalent mutations and geographic differences. This descriptive approach ensures clarity and allows assessment of regional patterns and prevalence without statistical pooling or use of consistency measures, such as I².
Given the descriptive nature of the included studies and the focus on genetic variant reporting, no effect estimates, confidence intervals, or meta-analytic measures were calculated. Study-level variant data are presented in detail in tables to allow assessment of variant frequencies and regional patterns.
Results
Search results and study selection
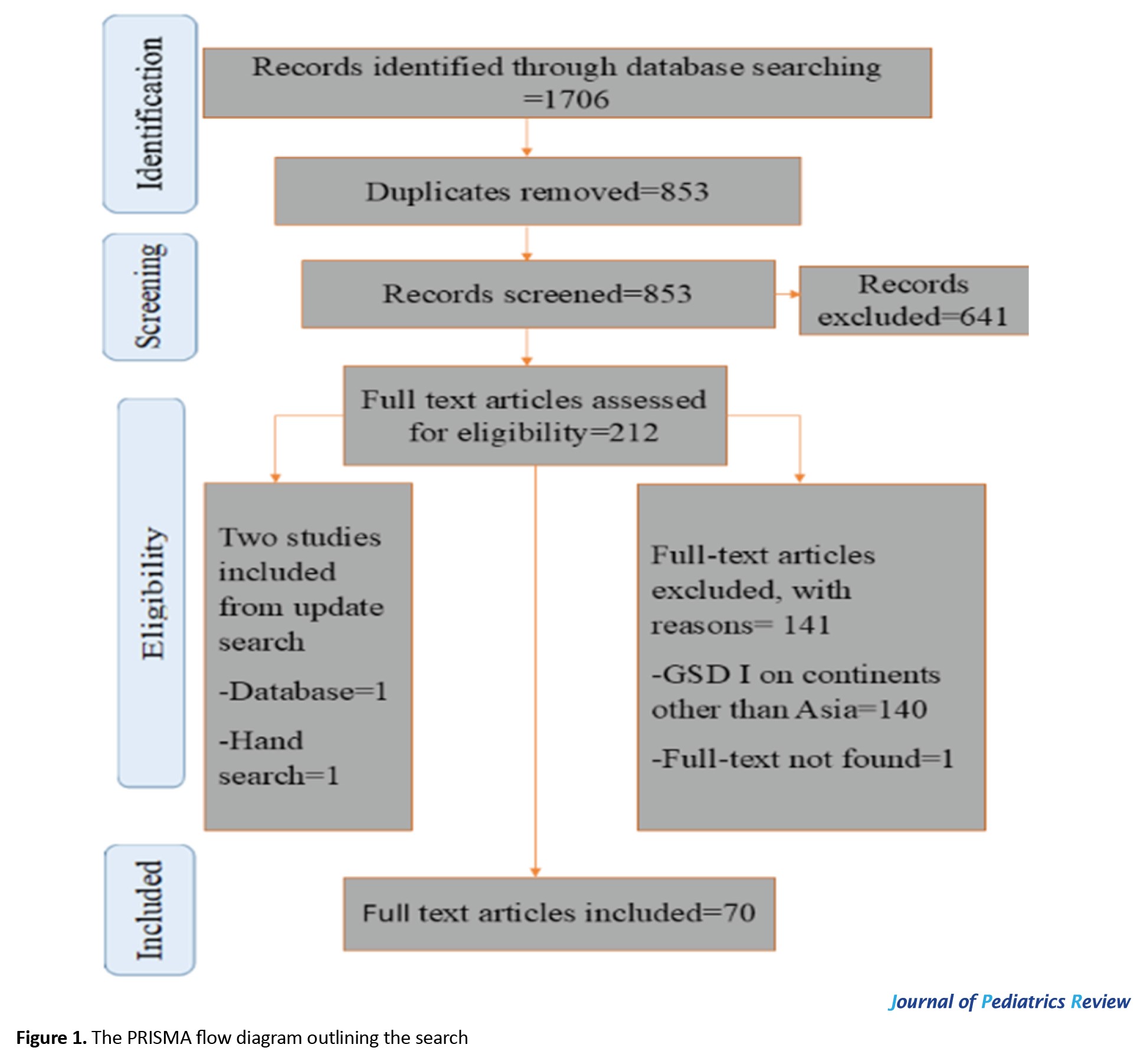
A total of 1,706 records were identified across the Web of Science, PubMed, Embase, Scopus, and ProQuest databases. After removing duplicates, 853 unique records remained. Screening of titles and abstracts excluded 641 studies that did not meet the inclusion criteria. The full texts of 211 articles were subsequently assessed for eligibility, resulting in the exclusion of 140 studies that did not focus on Asian populations, one study lacking a full text, and 29 studies for other reasons (e.g. non-human studies or insufficient data). An updated search and manual review added two additional studies, yielding a total of 70 studies included in this systematic review.
These included studies, published between 1995 and 2023, encompassed 680 patients from 14 countries and represented diverse ethnic groups and physiographic regions across Asia. The characteristics of the included studies, including country, patient numbers, disease subtypes, and mutation details, are summarized in Tables 1 [5-7, 9-11, 22, 26-58], 2 [3, 13-17, 60-65], 3 [63, 67-71], categorized by region and ethnicity.
A PRISMA flow diagram (Figure 1) illustrates the detailed study selection process, including screening, eligibility assessment, and reasons for exclusion at each stage.
Study characteristics
The included studies comprised a total of 70 publications, including case reports, case series, cross-sectional studies, and cohort studies, published between 1995 and 2023. Sample sizes varied widely, ranging from single-patient case reports to multicenter studies involving more than 100 patients. Overall, the studies included 680 patients diagnosed with glycogen storage disease type I, encompassing both GSD Ia (G6PC1-related) and GSD Ib (SLC37A4-related) subtypes.
Participants were patients of Asian origin from 14 countries across East Asia, West Asia, and South Asia. The primary outcomes assessed in all studies were the identification and frequency of pathogenic or likely pathogenic variants in the G6PC1 and/or SLC37A4 genes. Most studies employed molecular genetic techniques such as Sanger sequencing, next-generation sequencing, or whole-exome sequencing for variant detection. Funding sources and risk-of-bias assessments were not consistently reported across studies and were therefore not analyzed at the individual study level.
Due to the descriptive and genetic nature of the included studies, no interventions, comparators, or follow-up periods were applicable. Detailed characteristics of each study, including country, study design, sample size, disease subtype, and identified variants, are summarized in Tables 1 [5-7, 9-11, 22, 26-58], 2 [3, 13-17, 60-65], 3 [63, 67-73].
Across the 70 included studies, qualitative assessment indicated that most studies employed validated molecular genetic methods for variant detection. However, smaller case reports or series from underrepresented regions may have introduced selection bias, and some studies lacked complete details on consanguinity or patient characteristics. These considerations were accounted for in the interpretation of variant distribution and frequency patterns.
Distribution and relative frequencies of G6PC and SLC37A4 gene variants
Tables 4, 5, and 6 present a comprehensive summary of the various G6PC gene variants and their distribution across different regions of Asia, while Tables 7, 8, and 9 present SLC37A4 gene variants.
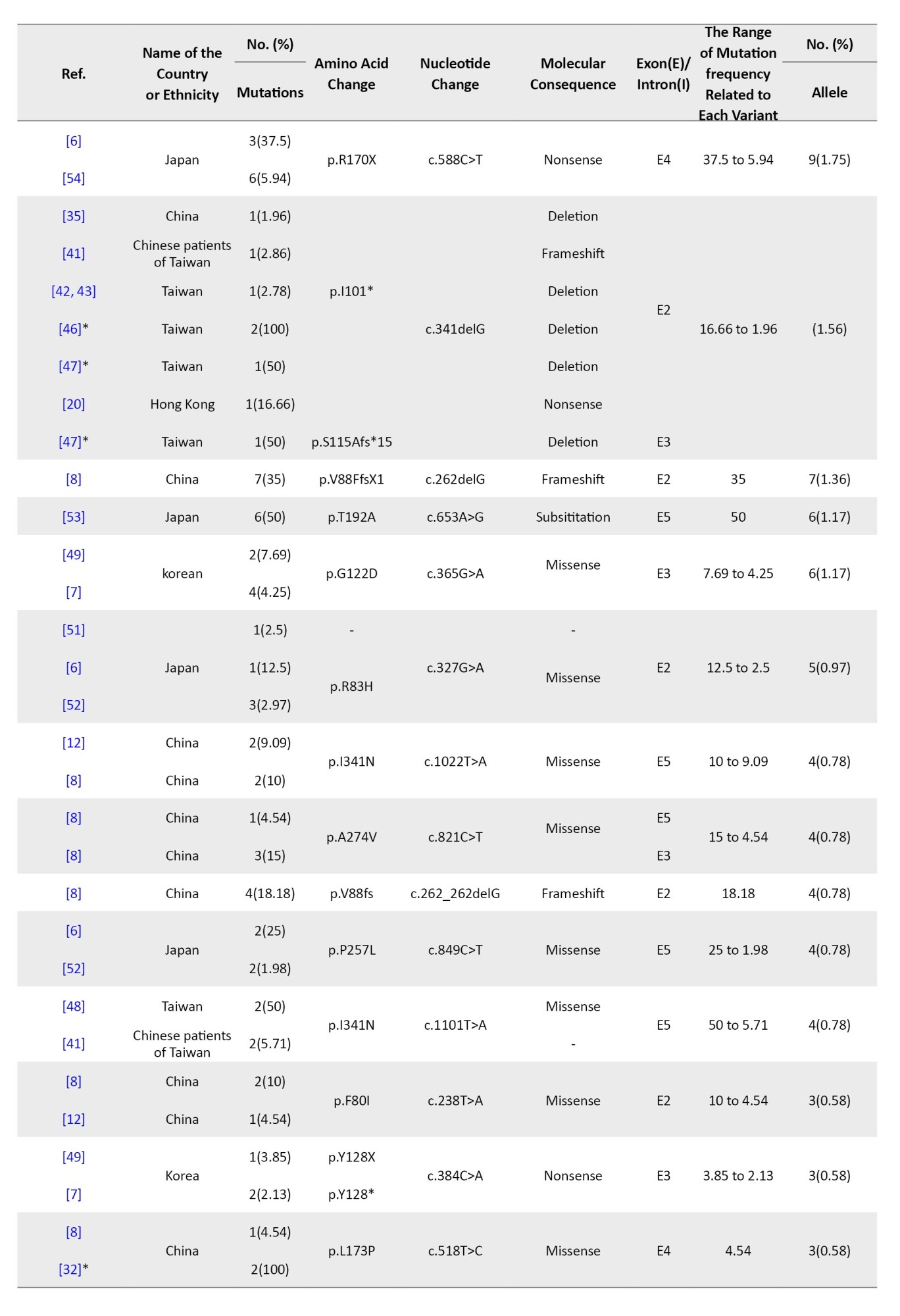
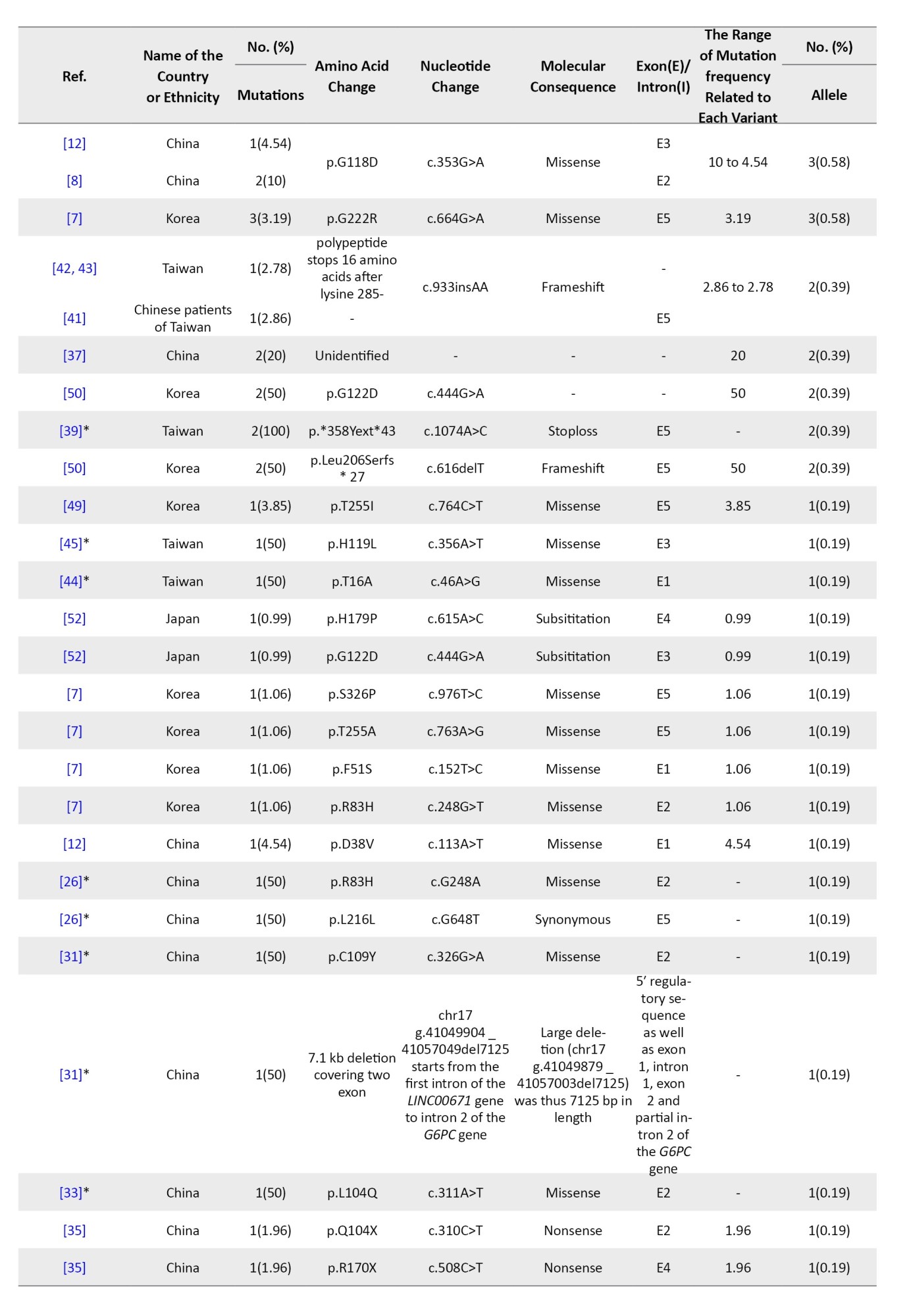
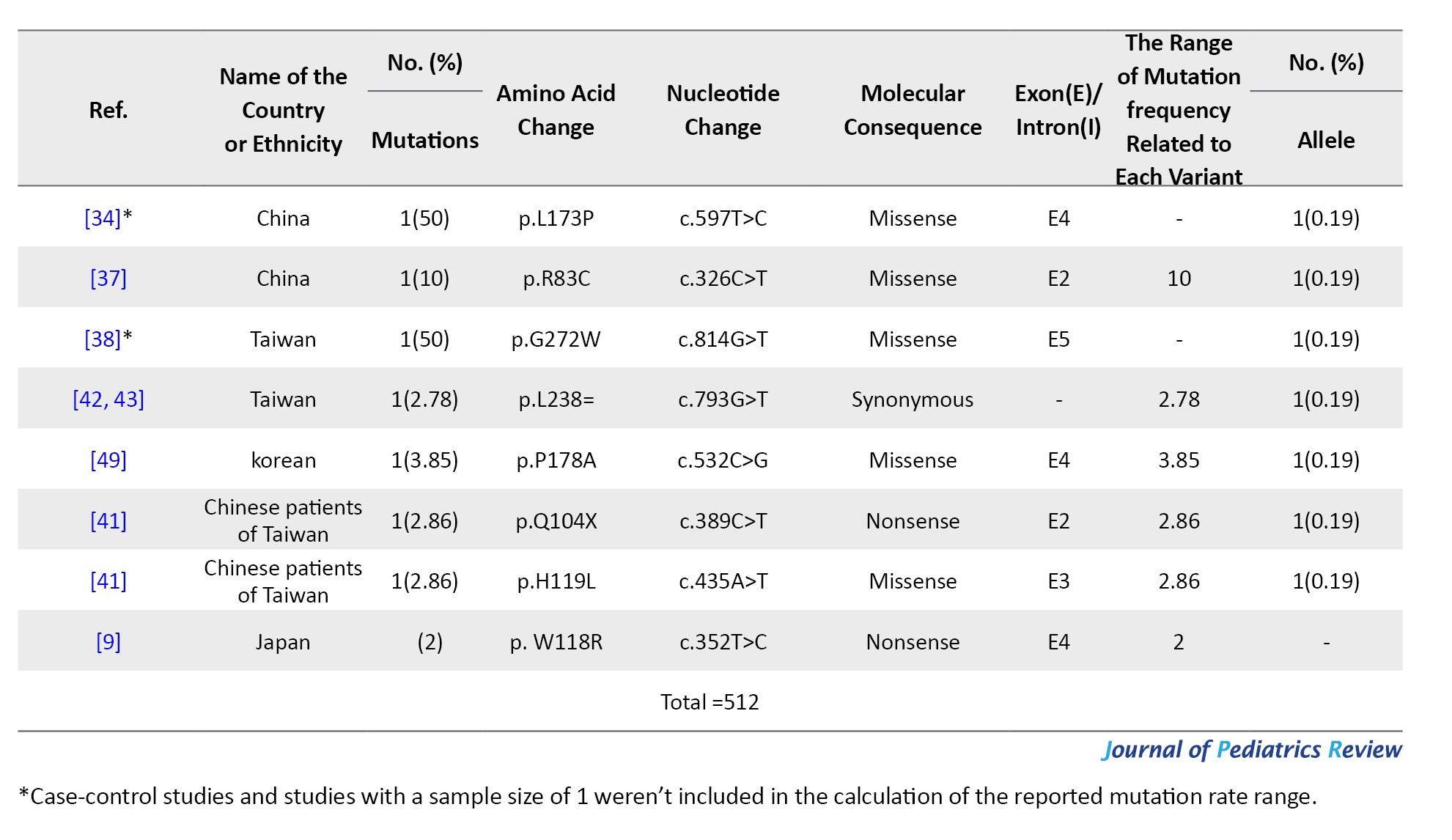
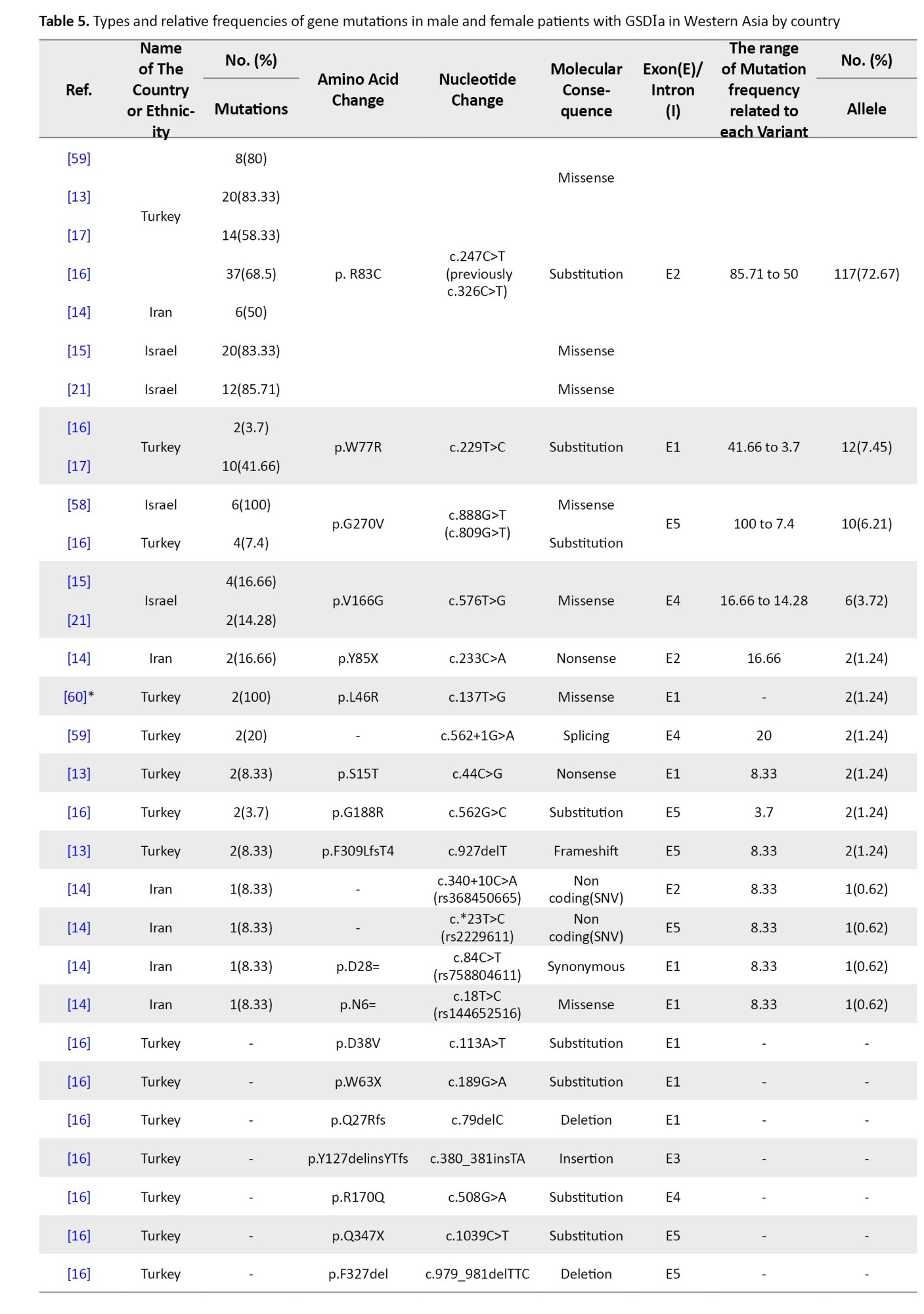
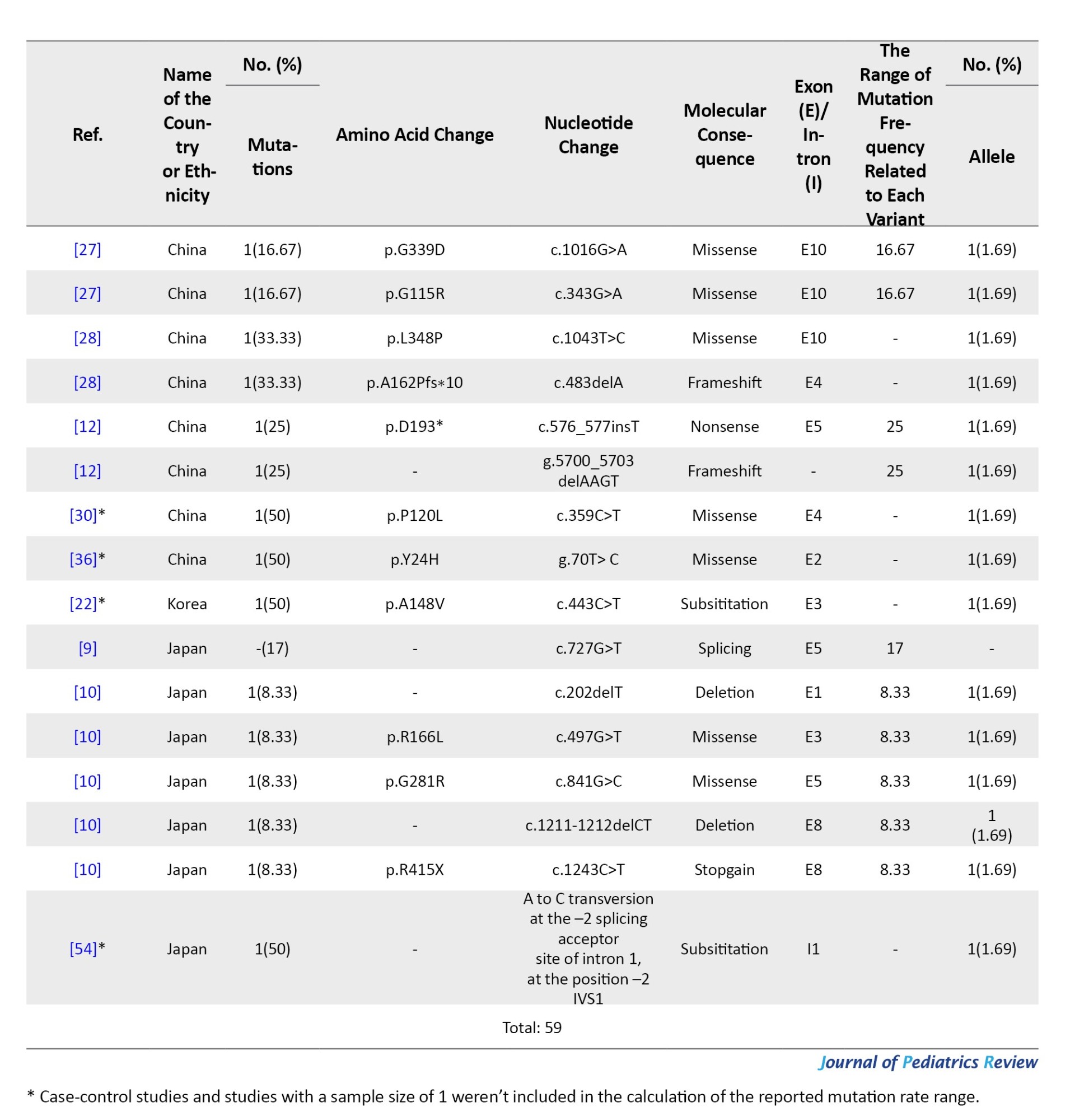
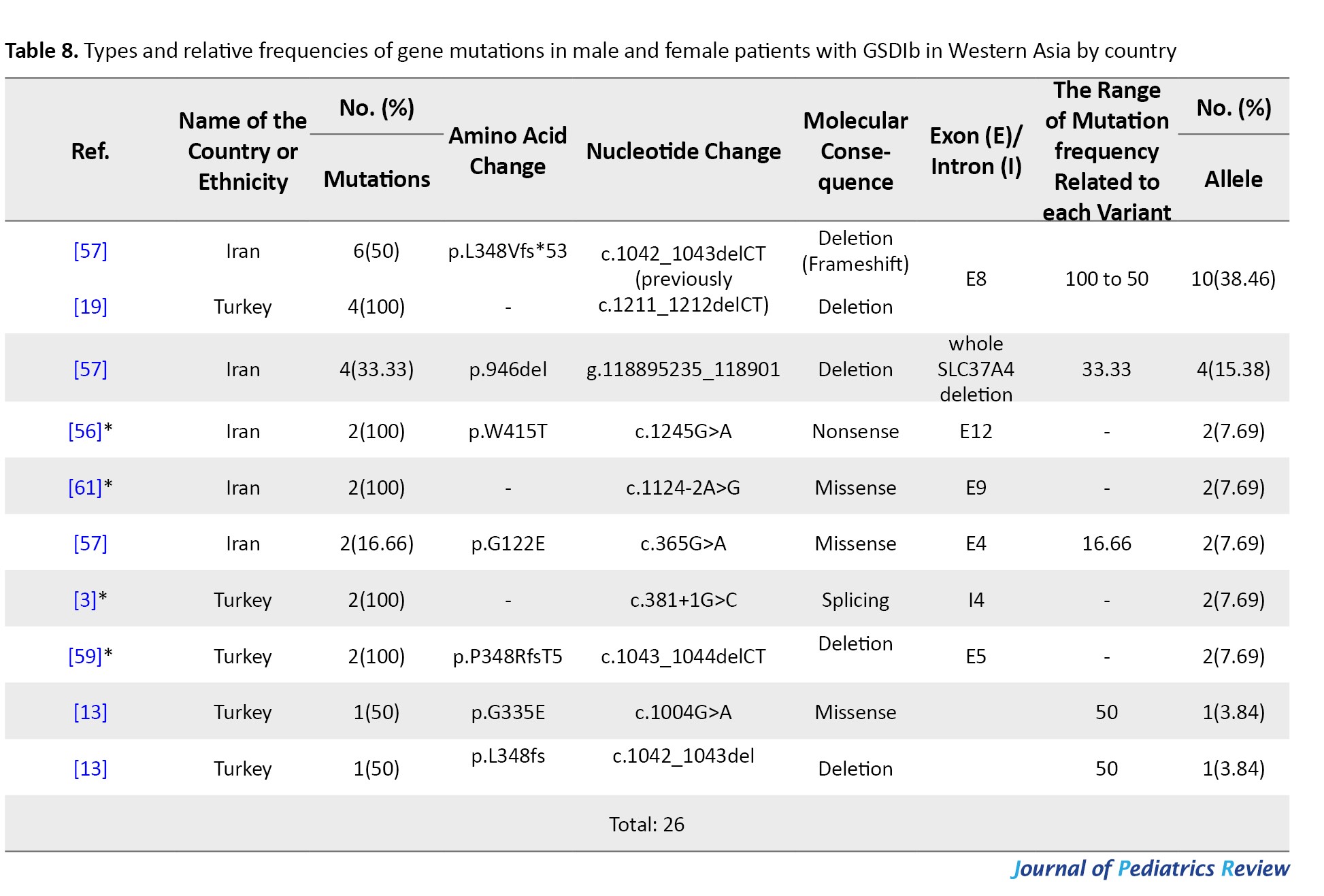
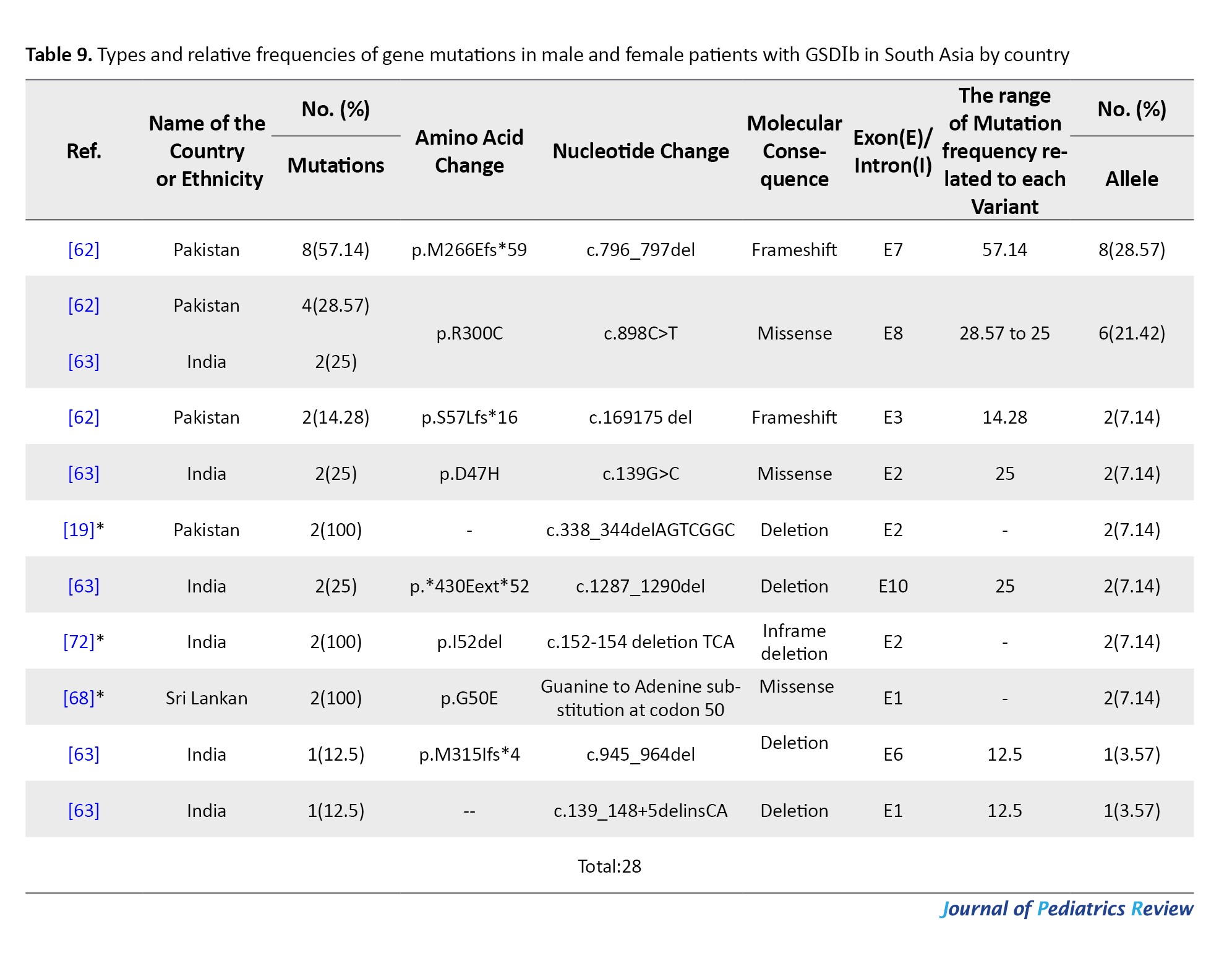
These tables detail the frequency of each variant type by country and ethnic group, based on individual study data. The total variant frequency for each gene was calculated by dividing the number of occurrences of each variant by the total number of reported variants associated with G6PC or SLC37A4. The column titled “variant frequency related to each study (%)” incorporates data from Tables 1, 2, and 3 for each respective country or ethnicity. The reported values reflect a range of variant frequencies observed in various populations, excluding those derived from case reports.
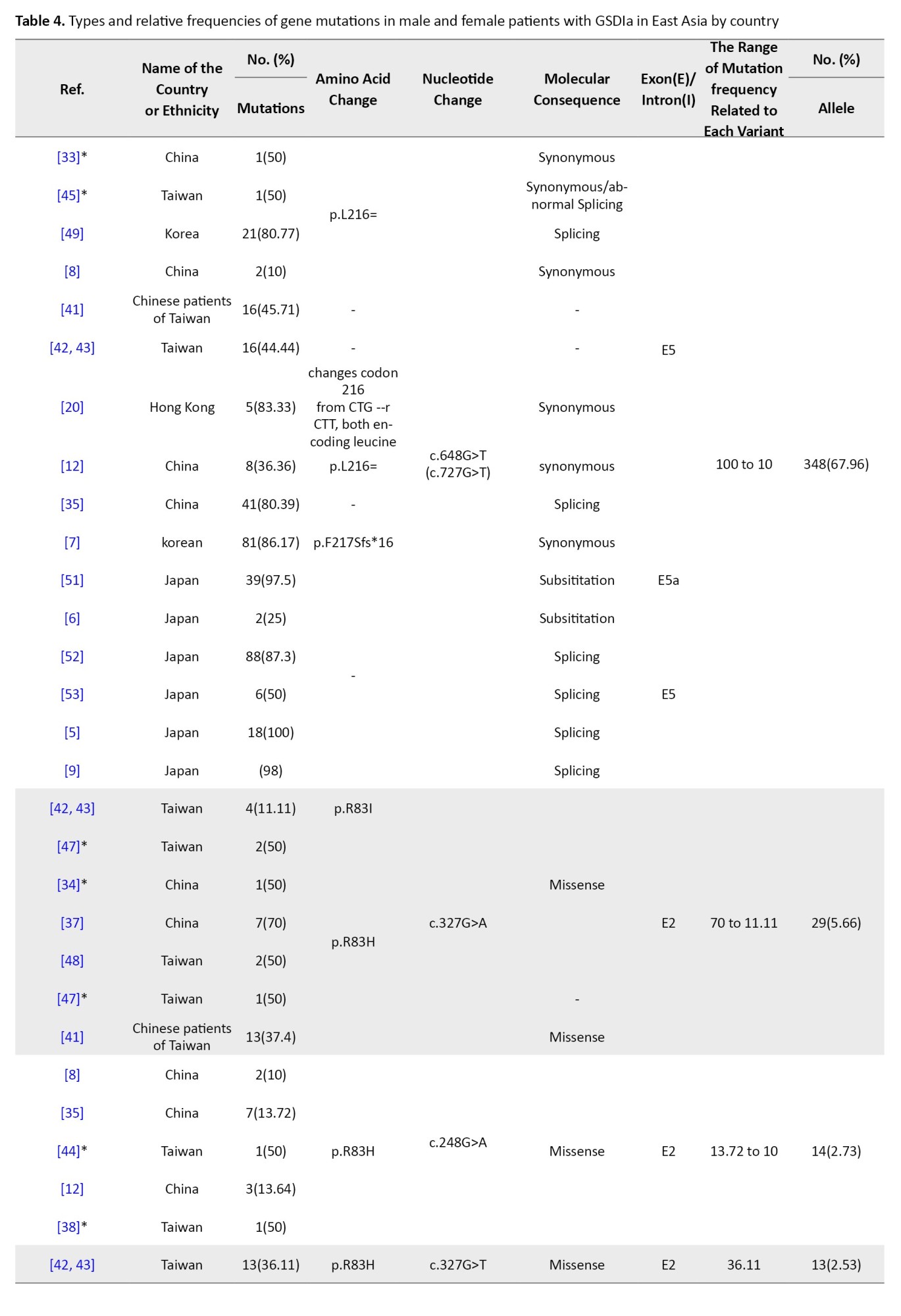
East Asia
East Asia includes China (with Hong Kong, Macau, and Tibet), Japan, Mongolia, North Korea, South Korea, and Taiwan. From 41 studies, 860 alleles were analyzed, identifying 571 mutated alleles and 83 unique mutations. Of these, 33 unique mutations were associated with the SLC37A4 gene and 50 with the G6PC gene, yielding an overall mutation detection rate of 66.39% (Table 1).
G6Pase gene variants in East Asia
A total of 27 studies across China, Japan, Taiwan, Korea, and Hong Kong investigated G6PC mutations in 245 patients. In total, 50 different mutations were found among 512 mutant alleles. The majority were missense (45), followed by nonsense (9), splice site (7), frameshift (5), and other types. Mutations clustered mostly in exons 2, 3, 4, and 5 (Table 1).
The most prevalent mutation was c.648G>T, representing 67.96% of mutant alleles, and it was widely observed in Japan (153 alleles), Korea (102), China (68), Taiwan (17), and Hong Kong (5). This was followed by c.327G>A (p.R83H) with 5.66% frequency, especially in Chinese and Taiwanese patients. Other notable variants included c.248G>A, c.327G>T, and c.588T>G (p.R170X), each ranging from approximately 1.7% to 2.7% (Table 4).
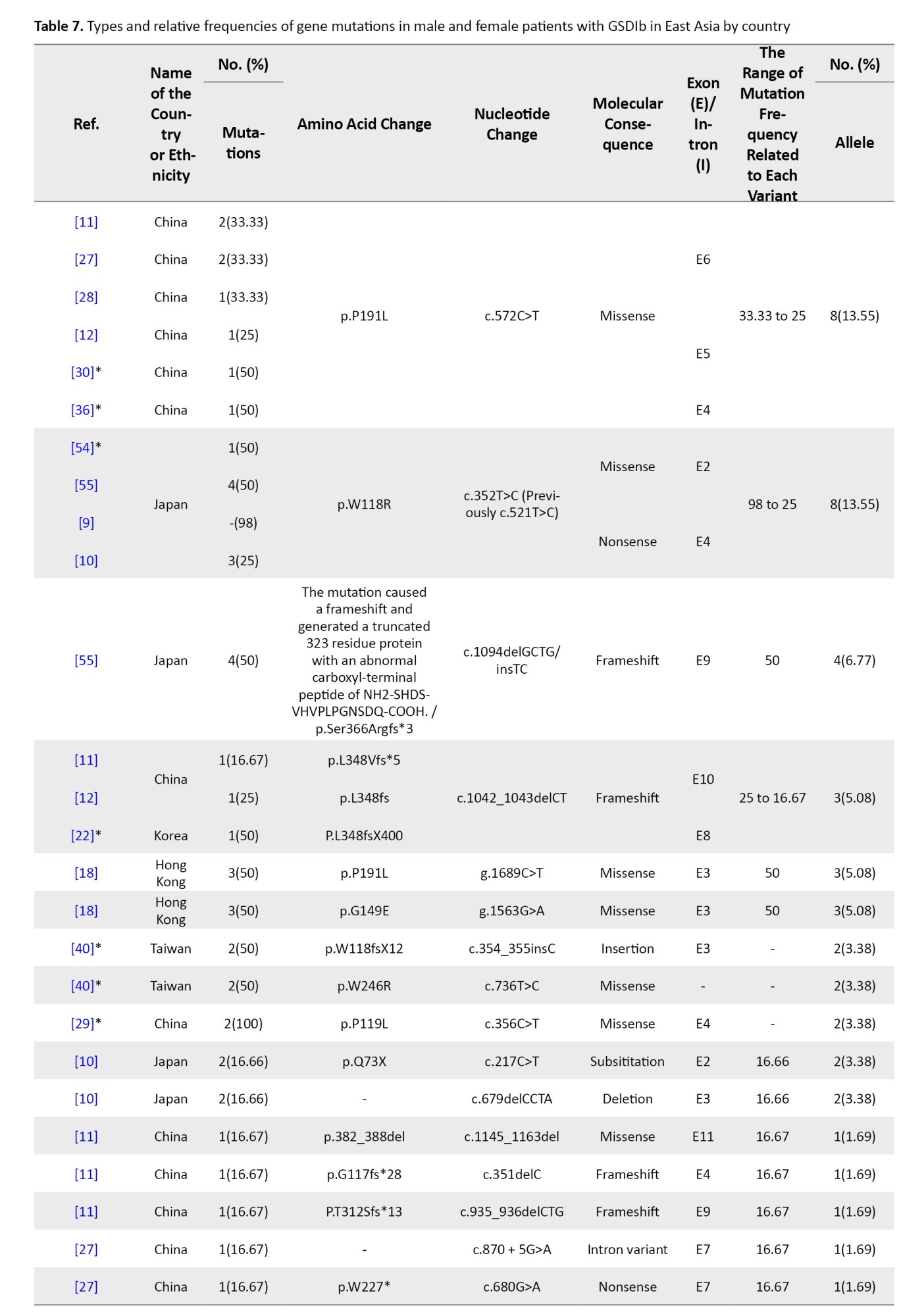
SLC37A4 gene variants in East Asia
Twelve studies documented 33 SLC37A4 mutations across 59 alleles. The most common were in exons 4, 3, 5, and 10. A splice-site mutation in intron 1 (IVS1 -2 A>C) was also identified. The most frequent variant in China was c.572C>T (p.P191L) (13.55%), while c.521C>T (p.W118R) dominated in Japan (8.62%).
Another significant mutation, c.1094delGCTG/insTC (p.Ser366Argfs*3), produced a truncated protein and accounted for 6.77% of mutations. c.1042_1043delCT, a frameshift mutation in exon 8, was particularly common in China and Korea (5.08%).
Other recurrent variants included c.352T>C, g.1689C>T, c.679delCCTA, and c.935_936delCTG, all found at low to moderate frequencies across regional studies (Tables 1 and 3).
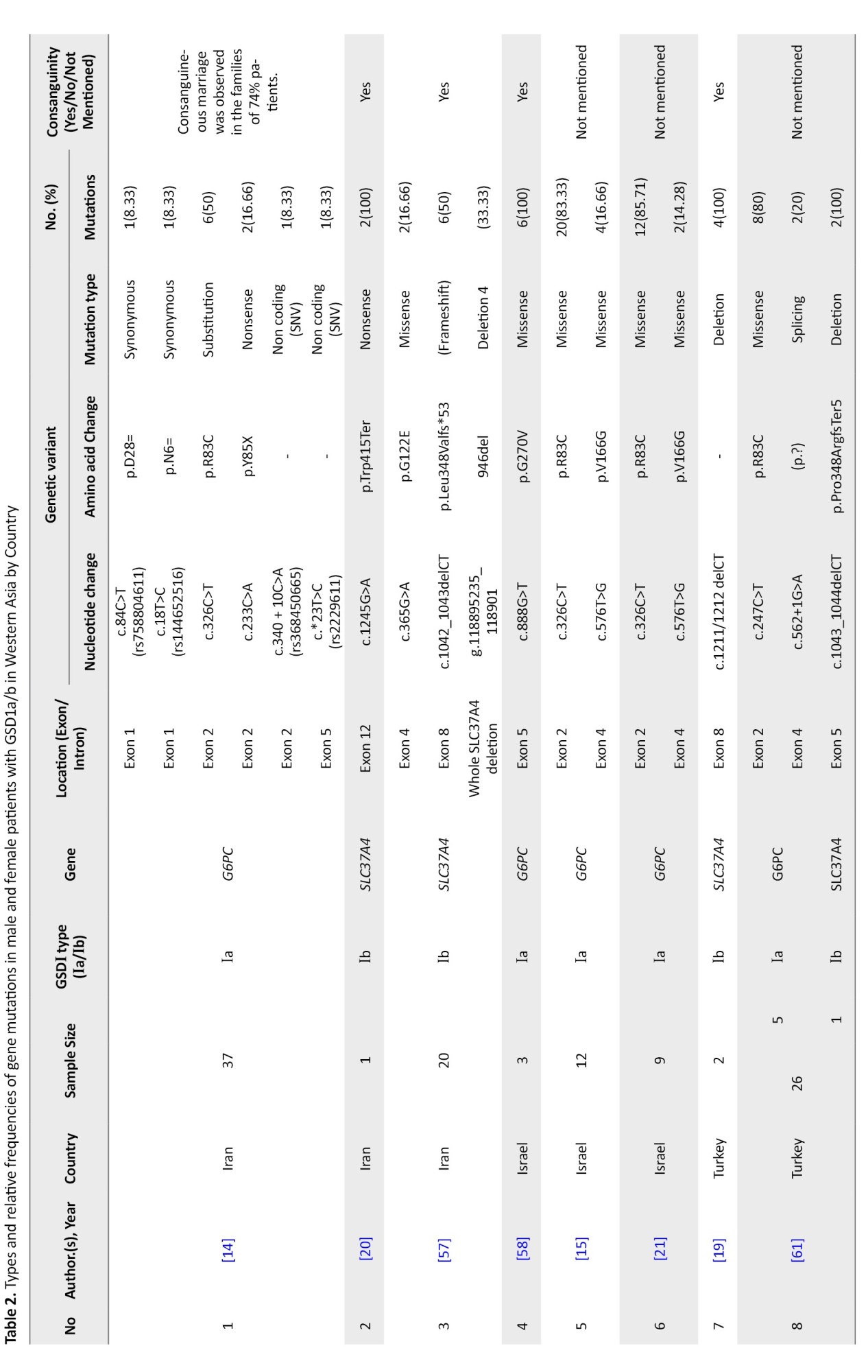
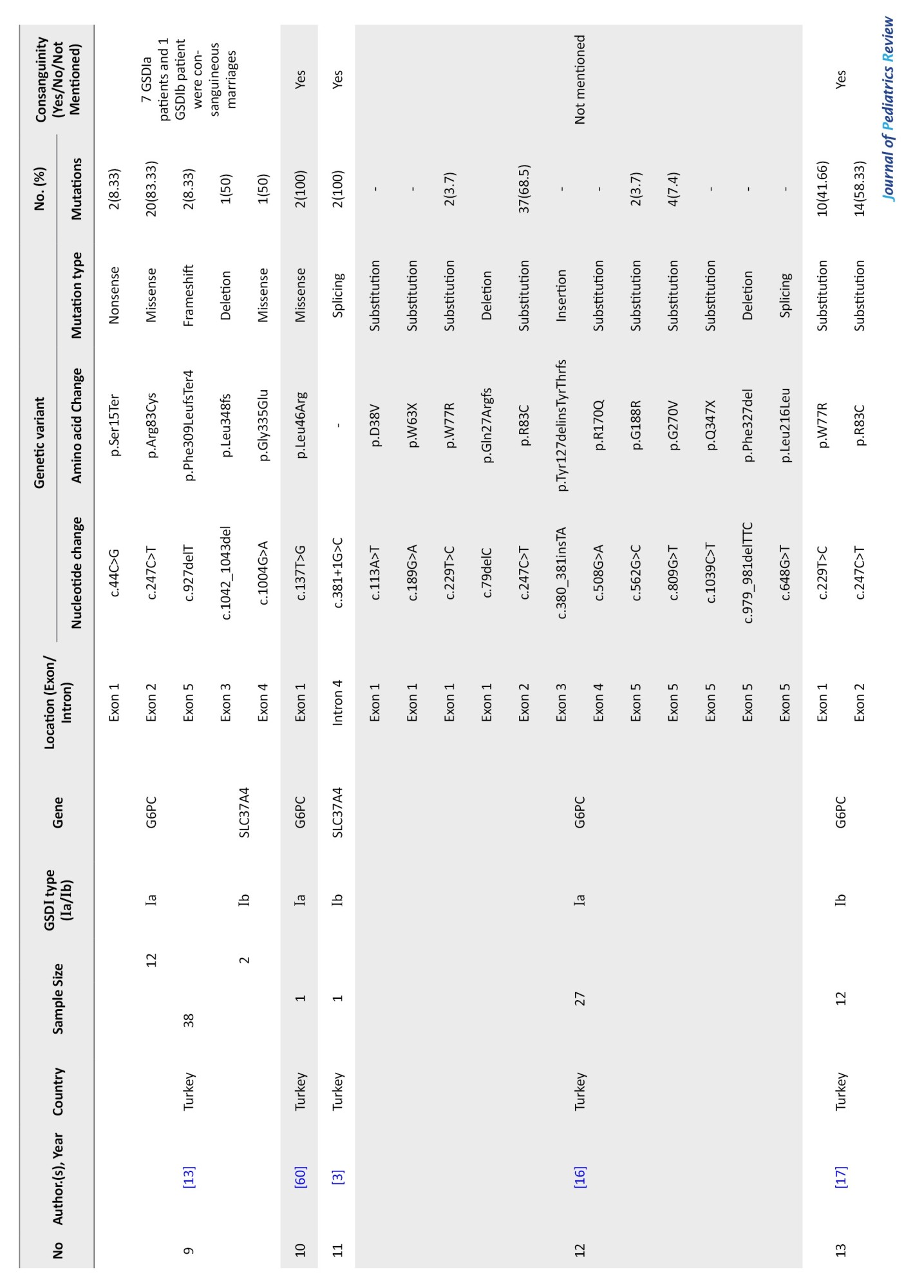
West Asia
Thirteen studies from Turkey, Iran, and Israel reported data on 190 patients, including 118 with GSD Ia and 28 with GSD Ib. Across these, 33 distinct variants were identified in 145 patients (Table 2).
G6PC gene variants in West Asia
Research on G6PC gene variants in West Asia has been limited to three countries: Turkey, Iran, and Israel, despite the region comprising 20 countries, including Armenia, Azerbaijan, and Saudi Arabia.
Nine studies focused on G6PC mutations, reporting 24 distinct variants across 236 alleles, of which 161 were mutated (mutation rate: 68.22%). Israel showed the highest mutation detection rate (100%), followed by Turkey (92.1%) and Iran (16.21%). The most common mutation was c.247C>T (p.R83C), observed in 72.67% of cases and across all three countries. p.W77R (c.229T>C) was the second most frequent (7.45%), especially in Turkish cohorts. c.888G>T (p.G270V) ranked third (6.21%) and was also reported from both Turkey and Israel. Additional low-frequency variants included c.576T>G, c.809G>T, and several private or rare substitutions (Table 5).
SLC37A4 gene variants in West Asia
The mutation spectrum of the SLC37A4 gene has been documented in only two West Asian countries: Turkey and Iran. Six studies from Turkey and Iran reported 9 unique SLC37A4 variants in 27 patients. The overall mutation detection rate was 50%. Most patients (92.6%) came from consanguineous families.
The most frequent variant was c.1042_1043delCT (p.L348Vfs*53), found in 41.66% of cases, with 50–100% frequency across studies. In Iran, notable variants included g.118895235_118901del (946del) (16.66%) and c.1245G>A (p.W415T) (8.33%). In Turkey, c.381+1G>C and c.1043_1044delCT (p.P348Rfs*5) were found in smaller proportions (8.33% each) (Table 8).
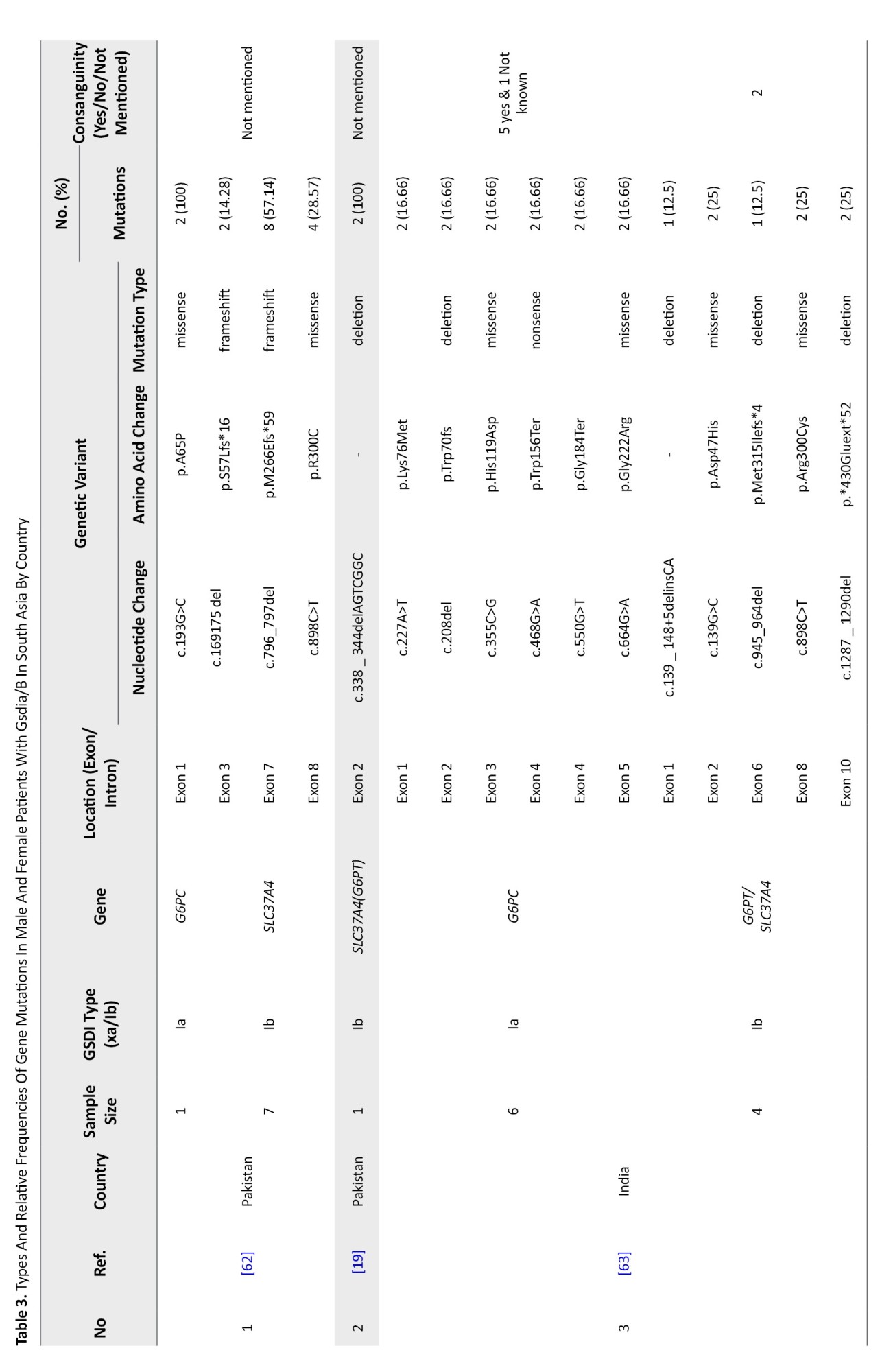
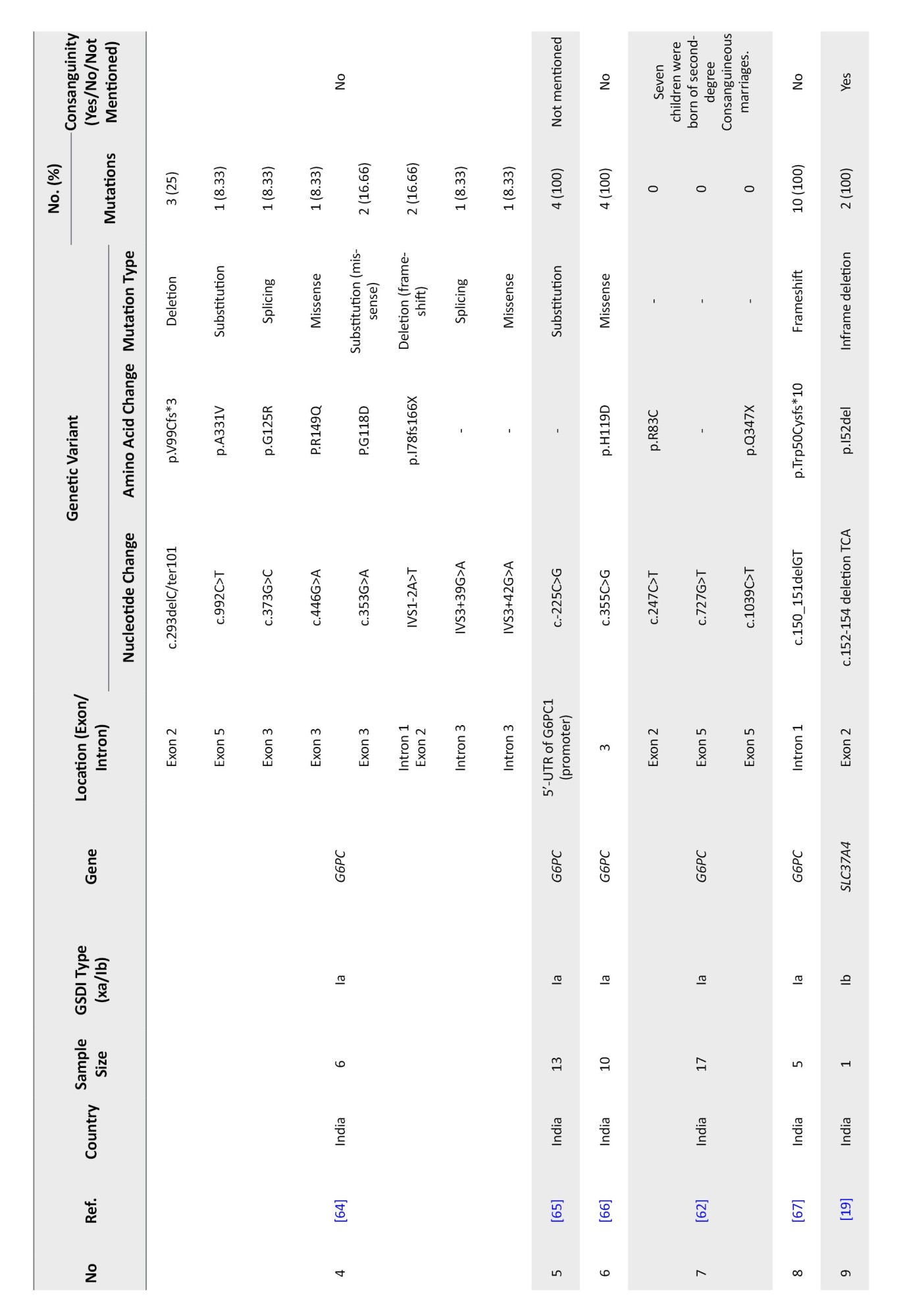
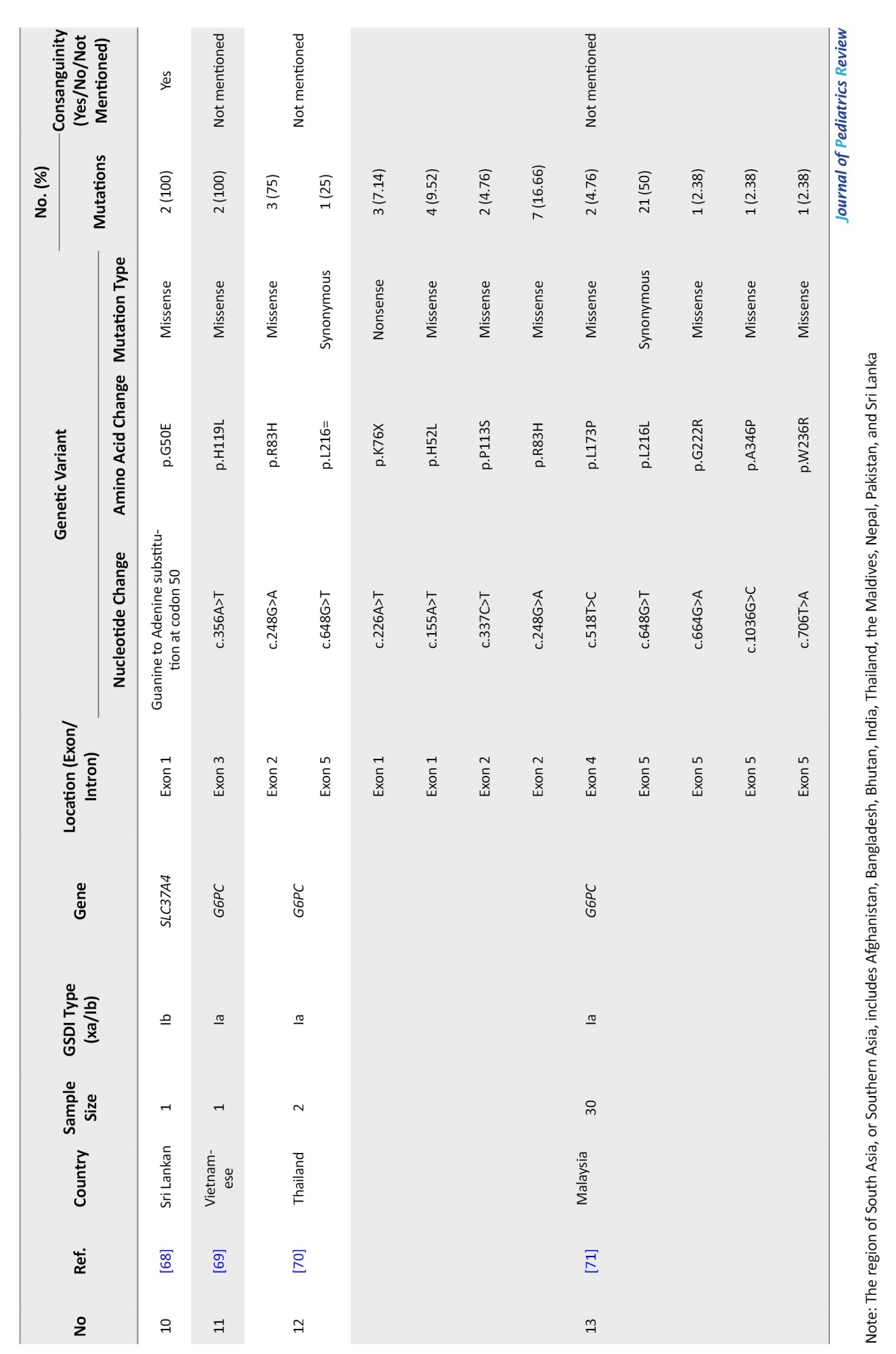
South Asia
South Asia encompasses Afghanistan, Bangladesh, Bhutan, India, Maldives, Nepal, Pakistan, and Sri Lanka. Thirteen studies involving 105 patients identified 39 distinct variants across 120 alleles—29 related to GSD Ia and 10 to GSD Ib. Consanguinity was reported in only 15 families (Table 3).
G6PC gene variants in South Asia
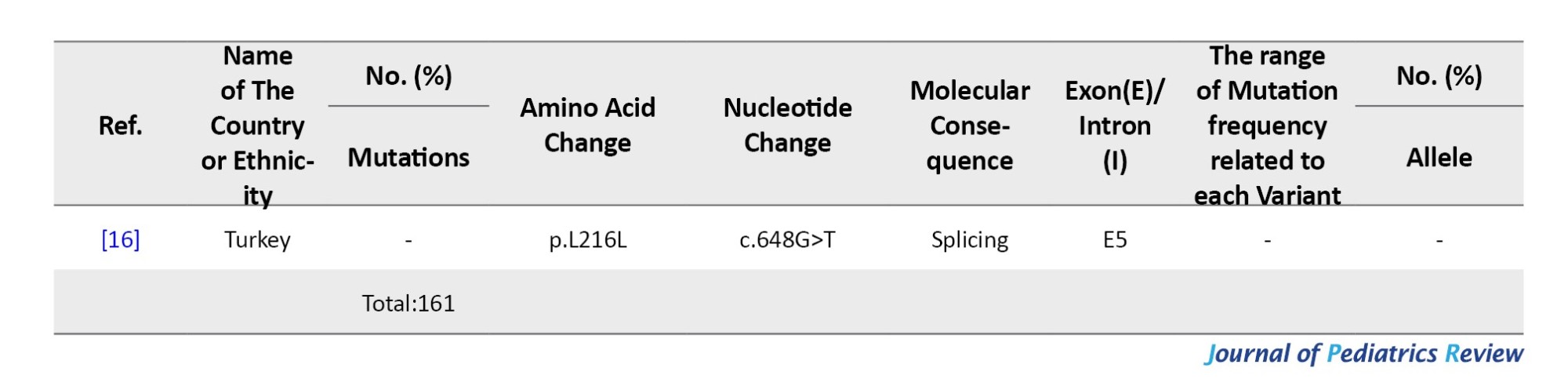
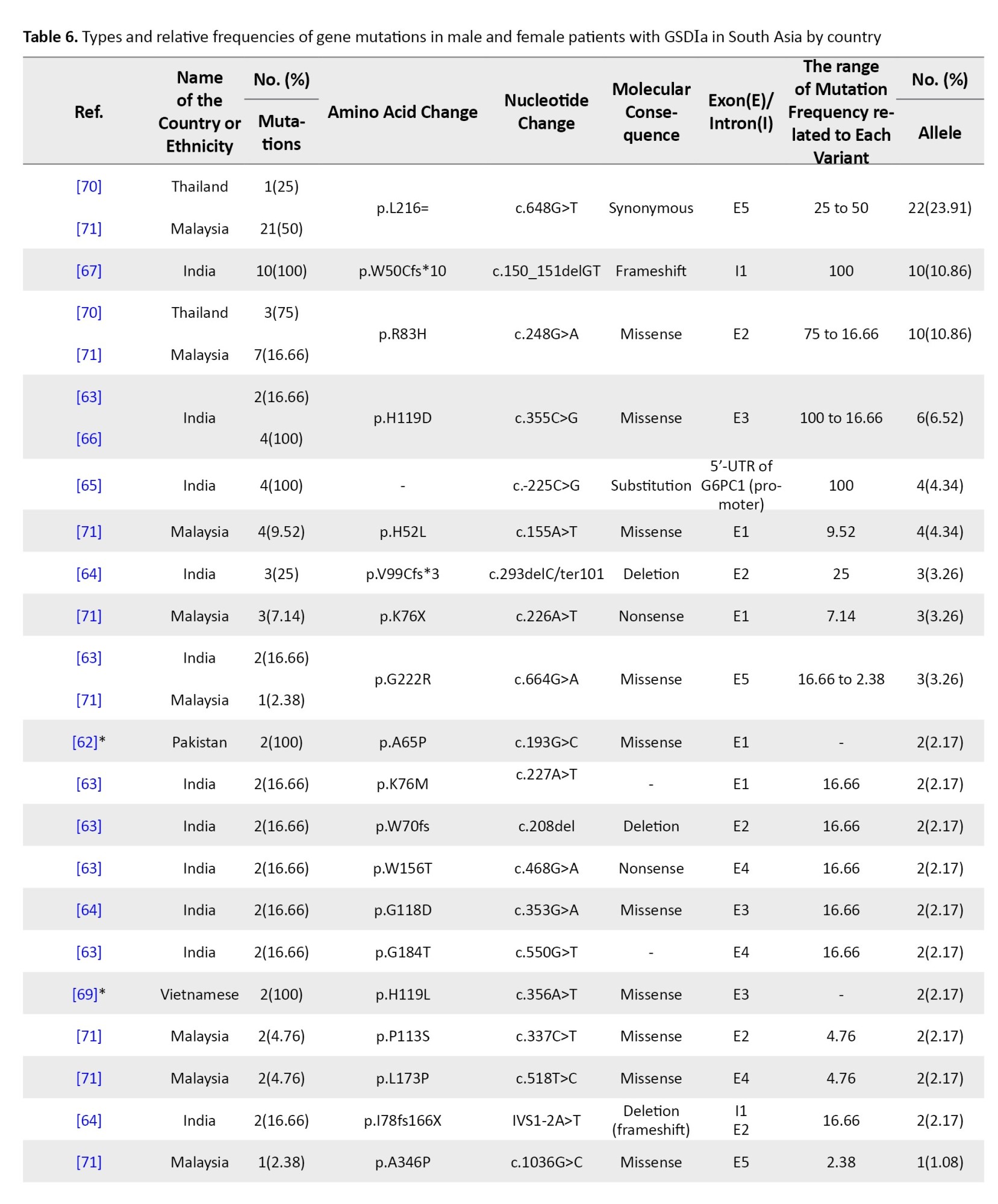
Between 2001 and 2022, 29 G6PC mutations were identified in Malaysia, Thailand, India, Pakistan, and Vietnam. India contributed the most data (57 patients, 19 variants). Malaysia reported 42 mutated alleles in 30 patients. Shared mutations included c.648G>T and c.248G>A in Malaysia and Thailand.
The most frequent mutation was c.648G>T (p.L216=), especially in Thailand and Malaysia (23.91%). In India, c.150_151delGT (p.W50Cfs*10) was most common (10.86%), followed by c.355C>G (p.H119D) (6.52%) and c.-225C>G (4.34%). c.248G>A was the third most common overall (10.86%). Mutations mainly clustered in exons 2, 3, and 5 (Table 6).
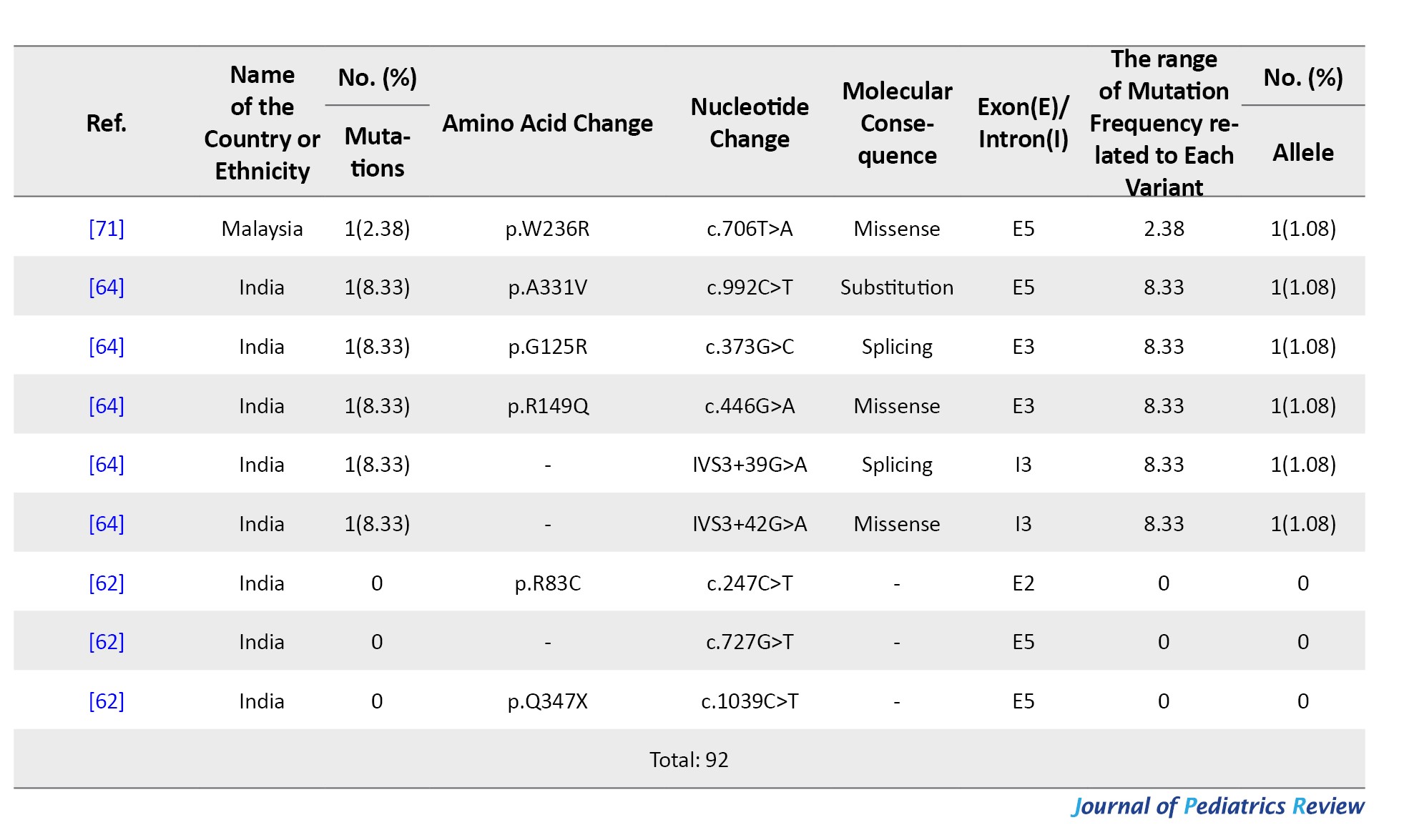
SLC37A4 gene variants in South Asia
From 2000 to 2022, five studies (14 patients) identified 10 SLC37A4 mutations across India, Pakistan, and Sri Lanka. A total of 28 alleles were analyzed with a 100% detection rate. Four patients were from consanguineous families.
Variants included 4 missense mutations, 2 frameshifts, 4 deletions, and 1 in-frame deletion. Most mutations were located in exons 1 and 2. The most common variant was c.796_797del (p.M266Efs*59) in Pakistan (28.57%), followed by c.898C>T (p.R300C) in India (21.42%). Other less frequent variants included c.169_175del (p.S57Lfs*16) and c.150G>A (p.G50E) (Table 9).
Discussion
This systematic review analyzed 70 studies, including 680 patients from 14 Asian countries, examining mutations in the G6PC1 and SLC37A4 genes among patients with GSD Ia and Ib. By organizing variants geographically across East, West, and South Asia, we identified both shared and region-specific mutation patterns. Key prevalent mutations included c.648G>T in East and South Asia, R83C and p.W77R in West Asia, and c.572C>T in China, providing strong evidence for regionally tailored genetic screening strategies.
These findings have clear clinical relevance: they guide healthcare providers and genetic counselors in accurate diagnosis, targeted molecular testing, and informed counseling, particularly in populations with high consanguinity and founder effects. The data from East Asia are particularly robust, while evidence from other regions remains limited, highlighting the need for further research in underrepresented populations. Overall, this review establishes a comprehensive genetic landscape of GSD I in Asia, supporting both clinical decision-making and future investigations into variant prevalence, regional distribution, and population-specific founder effects.
East Asia showed the highest mutation reporting, particularly from Japan, Korea, China, and Taiwan. The most frequent G6PC1 variant was c.648G>T (c.727G>T), representing 67.96% of all mutations in the region and reaching up to 92% in Korean patients and 81% in Japanese cohorts [5, 7, 9]. This variant causes a splicing error, resulting in a truncated G6Pase protein with approximately 18% enzymatic activity [5]. It appears to be a founder mutation, especially in Japanese populations. Other frequent East Asian mutations included c.327G>A (R83H) and c.248G>A, both missense mutations within exon 2, accounting for 5.6% and 3.1% of mutations respectively, especially among Chinese and Taiwanese populations [6]. These mutations were less common in Korea and Japan (Table 4).
In SLC37A4, East Asian patients commonly exhibited c.572C>T (P191L) and c.352T>C (W118R) (Table 7) [9, 10]. The former represented 23% of mutations in Chinese studies, while the latter was dominant in Japanese GSD Ib patients (17%) [6]. The c.1042_1043delCT deletion also appeared in 4.6% of alleles across Korean and Chinese patients [11, 12]. West Asia showed dominance of the c.247C>T (R83C) mutation in G6PC1, found in 72.67% of cases in studies from Turkey, Iran, and Israel [13-15]. In Iranian cohorts alone, it was reported in over 80% of patients. The p.W77R mutation followed with 11.3% frequency in Turkish patients, while c.888G>T (G270V) made up 6.2% of reported variants [16, 17].
For SLC37A4, the c.1042_1043delCT deletion was the most frequent in Iranian GSD Ib patients (64%), followed by g.118895235_118901del (10.5%) and c.1245G>A (W415T) (7.9%) [18-20].
South Asia displayed a broader mutation spectrum. The c.648G>T mutation appeared again, particularly in Malaysia (31%) and Thailand (27%). In India, the most frequent G6PC1 mutation was c.150_151delGT (W50Cfs*10), comprising 39.5% of cases [21]. Other regionally shared variants were c.248G>A (13%) and c.355C>G (H119D, 8.5%).
In SLC37A4, Pakistani and Indian patients showed frequent c.796_797del (M266Efs*59) and c.898C>T (R300C) mutations, accounting for 44.6% and 28.7% respectively in localized studies [22]. These mutations were rarely identified in other Asian regions.
Comparative data from Europe indicates that R83C, the dominant West Asian variant, is also prevalent in Mediterranean populations but nearly absent in East Asia. Conversely, c.648G>T, common in East and South Asia, is largely undetected in European cohorts. The c.327G>A variant, although present in Asia, occurs at significantly lower frequencies in Europe [23-25].
These findings support the design of regionally customized genetic screening panels. Including prevalent variants such as c.648G>T, R83H, and c.572C>T for East/South Asia and R83C, p.W77R for West Asia can significantly improve diagnostic yield. High consanguinity rates and population-specific founder effects further emphasize the value of population-based genetic approaches in GSD I diagnosis and counseling.
Conclusion
This systematic review highlights substantial genetic diversity in G6PC1 and SLC37A4 mutations among Asian patients with GSD I. Distinct mutation patterns across East, West, and South Asia underscore the necessity for region-specific genetic screening and tailored diagnostic strategies. Key prevalent variants, such as c.648G>T in East/South Asia and c.572C>T in China, enhance diagnostic accuracy and can guide targeted patient management.
These findings provide a comprehensive overview of the genetic landscape of GSD I in Asia, supporting clinical decision-making, informed genetic counseling, and public health planning, particularly in populations with high consanguinity or founder effects. Furthermore, the study highlights gaps in data from underrepresented regions and emphasizes the need for future research employing advanced sequencing approaches, larger cohorts, and inclusive population sampling to better characterize variant frequencies and regional patterns.
Overall, this review underscores the critical role of genetic testing in improving patient outcomes and lays a foundation for personalized medicine approaches in GSD I across diverse Asian populations.
Limitations
This study provides valuable insights into GSD I mutations by analyzing genetic variations across different geographic regions in Asia, the largest continent with a population of approximately 4.7 billion. However, several limitations should be acknowledged. At the study and outcome levels, many included studies had small sample sizes and limited representation from certain countries, which may affect the accuracy and generalizability of variant frequency estimates. At the review level, despite conducting a comprehensive systematic search across multiple databases, some studies may have been missed due to language restrictions, incomplete reporting, or limited accessibility, potentially introducing reporting bias. Furthermore, our analysis included only 32 of 48 Asian countries, encompassing 680 individuals, which may not fully capture the continent’s ethnic and geographic diversity. These limitations highlight the need for broader analyses, particularly in underrepresented populations, and suggest that future studies could benefit from using whole-exome sequencing or other high-throughput genetic approaches to provide a more complete picture of genetic variation in GSD I across Asia.
Ethical Considerations
Compliance with ethical guidelines
The study protocol was approved by the Research Ethics Committee of Isfahan University of Medical Sciences, Isfahan, Iran (Code: IR.ARI.MUI.REC.1403.155).
Funding
This work was supported by Isfahan University of Medical Sciences, Isfahan, Iran (Grant No.: 1402262).
Authors contributions
All authors contributed equally to the conception and design of the study, data collection and analysis, interception of the results and drafting of the manuscript. Each author approved the final version of the manuscript for submission.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors acknowledge the support of the Vice Chancellor for Research at Isfahan University of Medical Sciences, Isfahan, iRAN.
References
Supplementary Material S1
PubMed
PICo
Population= (“Glycogen storage disease type Ia”[tiab] OR “Glycogen storage disease type Ib”[tiab] OR “Von Gierke disease”[tiab] OR “Type I Glycogen storage disease”[tiab] OR “Type Ia Glycogen storage disease”[tiab] OR “Type Ib Glycogen storage disease”[tiab] OR “Glycogen storage disease type 1”[tiab] OR “Glycogen storage disease type 1a”[tiab] OR “Glycogen storage disease type 1b”[tiab] OR “Type 1 Glycogen storage disease”[tiab] OR “Type 1a Glycogen storage disease”[tiab] OR “Type 1b Glycogen storage disease”[tiab] OR “Glycogen storage disorder type I”[tiab] OR “Glycogen storage disorder type Ia”[tiab] OR “Glycogen storage disorder type Ib”[tiab] OR “Glycogen storage disorder type 1”[tiab] OR “Glycogen storage disorder type 1a”[tiab] OR “Glycogen storage disorder type 1b”[tiab] OR “Type I Glycogen storage disorder”[tiab] OR “Type Ia Glycogen storage disorder”[tiab] OR “Type Ib Glycogen storage disorder”[tiab] OR “Type 1 Glycogen storage disorder”[tiab] OR “Type 1a Glycogen storage disorder”[tiab] OR “Type 1b Glycogen storage disorder”[tiab] OR “Gierke Disease”[tiab] OR (Disease[tiab] AND Gierke[tiab]) OR “Gierke’s Disease”[tiab] OR (Disease[tiab] AND Gierke’s[tiab]) OR “Gierkes Disease”[tiab] OR “Glucose-6-Phosphatase Deficiency”[tiab] OR (Deficienc*[tiab] AND “Glucose-6-Phosphatase”[tiab]) OR “Glucose 6 Phosphatase Deficiency”[tiab] OR “Glucose-6-Phosphatase Deficienc*”[tiab] OR “Glycogen Storage Disease 1”[tiab] OR “GSD I”[tiab] OR “Glycogenosis 1”[tiab] OR “Hepatorenal Glycogen Storage Disease”[tiab] OR “von Gierke Disease”[tiab] OR (Disease[tiab] AND “von Gierke”[tiab]) OR “von Gierke’s Disease”[tiab] OR (Disease[tiab] AND “von Gierke’s”[tiab]) OR “von Gierkes Disease”[tiab] OR (Deficiency[tiab] AND Glucosephosphatase[tiab]) OR (Deficiencies[tiab] AND Glucosephosphatase[tiab]) OR “Glucosephosphatase Deficienc*”[tiab])
Phenomenon of interest= (“Next-generation sequencing”[tiab] OR NGS[tiab] OR “Whole exome sequencing”[tiab] OR WES[tiab] OR (Sequencing[tiab] AND Exome[tiab]) OR “Whole Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Whole Transcriptome”[tiab]) OR (“Transcriptome Sequencing”[tiab] AND Whole[tiab]) OR “Complete Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Complete Transcriptome”[tiab]) OR (“Transcriptome Sequencing*”[tiab] AND Complete[tiab]) OR “Whole Exome Sequencing”[tiab] OR (“Exome Sequencing”[tiab] AND Whole[tiab]) OR (Sequencing[tiab] AND “Whole Exome”[tiab]) OR “Complete Exome Sequencing*”[tiab] OR (“Exome Sequencing*”[tiab] AND Complete[tiab]) OR (Sequencing[tiab] AND “Complete Exome”[tiab]) OR variants[tiab] OR “variants types”[tiab] OR “variants Abundance”[tiab] OR “variants frequency”[tiab] OR Variation[tiab] OR Mutation*[tiab])
Context=-
(“Glycogen storage disease type Ia”[tiab] OR “Glycogen storage disease type Ib”[tiab] OR “Von Gierke disease”[tiab] OR “Type I Glycogen storage disease”[tiab] OR “Type Ia Glycogen storage disease”[tiab] OR “Type Ib Glycogen storage disease”[tiab] OR “Glycogen storage disease type 1”[tiab] OR “Glycogen storage disease type 1a”[tiab] OR “Glycogen storage disease type 1b”[tiab] OR “Type 1 Glycogen storage disease”[tiab] OR “Type 1a Glycogen storage disease”[tiab] OR “Type 1b Glycogen storage disease”[tiab] OR “Glycogen storage disorder type I”[tiab] OR “Glycogen storage disorder type Ia”[tiab] OR “Glycogen storage disorder type Ib”[tiab] OR “Glycogen storage disorder type 1”[tiab] OR “Glycogen storage disorder type 1a”[tiab] OR “Glycogen storage disorder type 1b”[tiab] OR “Type I Glycogen storage disorder”[tiab] OR “Type Ia Glycogen storage disorder”[tiab] OR “Type Ib Glycogen storage disorder”[tiab] OR “Type 1 Glycogen storage disorder”[tiab] OR “Type 1a Glycogen storage disorder”[tiab] OR “Type 1b Glycogen storage disorder”[tiab] OR “Gierke Disease”[tiab] OR (Disease[tiab] AND Gierke[tiab]) OR “Gierke’s Disease”[tiab] OR (Disease[tiab] AND Gierke’s[tiab]) OR “Gierkes Disease”[tiab] OR “Glucose-6-Phosphatase Deficiency”[tiab] OR (Deficienc*[tiab] AND “Glucose-6-Phosphatase”[tiab]) OR “Glucose 6 Phosphatase Deficiency”[tiab] OR “Glucose-6-Phosphatase Deficienc*”[tiab] OR “Glycogen Storage Disease 1”[tiab] OR “GSD I”[tiab] OR “Glycogenosis 1”[tiab] OR “Hepatorenal Glycogen Storage Disease”[tiab] OR “von Gierke Disease”[tiab] OR (Disease[tiab] AND “von Gierke”[tiab]) OR “von Gierke’s Disease”[tiab] OR (Disease[tiab] AND “von Gierke’s”[tiab]) OR “von Gierkes Disease”[tiab] OR (Deficiency[tiab] AND Glucosephosphatase[tiab]) OR (Deficiencies[tiab] AND Glucosephosphatase[tiab]) OR “Glucosephosphatase Deficienc*”[tiab]) AND (“Next-generation sequencing”[tiab] OR NGS[tiab] OR “Whole exome sequencing”[tiab] OR WES[tiab] OR (Sequencing[tiab] AND Exome[tiab]) OR “Whole Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Whole Transcriptome”[tiab]) OR (“Transcriptome Sequencing”[tiab] AND Whole[tiab]) OR “Complete Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Complete Transcriptome”[tiab]) OR (“Transcriptome Sequencing*”[tiab] AND Complete[tiab]) OR “Whole Exome Sequencing”[tiab] OR (“Exome Sequencing”[tiab] AND Whole[tiab]) OR (Sequencing[tiab] AND “Whole Exome”[tiab]) OR “Complete Exome Sequencing*”[tiab] OR (“Exome Sequencing*”[tiab] AND Complete[tiab]) OR (Sequencing[tiab] AND “Complete Exome”[tiab]) OR variants[tiab] OR “variants types”[tiab] OR “variants Abundance”[tiab] OR “variants frequency”[tiab] OR Variation[tiab] OR Mutation*[tiab])
Embase
(“Glycogen storage disease type Ia”:ti,ab OR “Glycogen storage disease type Ib”:ti,ab OR “Von Gierke disease”:ti,ab OR “Type I Glycogen storage disease”:ti,ab OR “Type Ia Glycogen storage disease”:ti,ab OR “Type Ib Glycogen storage disease”:ti,ab OR “Glycogen storage disease type 1”:ti,ab OR “Glycogen storage disease type 1a”:ti,ab OR “Glycogen storage disease type 1b”:ti,ab OR “Type 1 Glycogen storage disease”:ti,ab OR “Type 1a Glycogen storage disease”:ti,ab OR “Type 1b Glycogen storage disease”:ti,ab OR “Glycogen storage disorder type I”:ti,ab OR “Glycogen storage disorder type Ia”:ti,ab OR “Glycogen storage disorder type Ib”:ti,ab OR “Glycogen storage disorder type 1”:ti,ab OR “Glycogen storage disorder type 1a”:ti,ab OR “Glycogen storage disorder type 1b”:ti,ab OR “Type I Glycogen storage disorder”:ti,ab OR “Type Ia Glycogen storage disorder”:ti,ab OR “Type Ib Glycogen storage disorder”:ti,ab OR “Type 1 Glycogen storage disorder”:ti,ab OR “Type 1a Glycogen storage disorder”:ti,ab OR “Type 1b Glycogen storage disorder”:ti,ab OR “Gierke Disease”:ti,ab OR (Disease:ti,ab AND Gierke:ti,ab) OR “Gierkes Disease”:ti,ab OR “Glucose-6-Phosphatase Deficiency”:ti,ab OR (Deficienc*:ti,ab AND “Glucose-6-Phosphatase”:ti,ab) OR “Glucose 6 Phosphatase Deficiency”:ti,ab OR “Glucose-6-Phosphatase Deficienc*”:ti,ab OR “Glycogen Storage Disease 1”:ti,ab OR “GSD I”:ti,ab OR “Glycogenosis 1”:ti,ab OR “Hepatorenal Glycogen Storage Disease”:ti,ab OR “von Gierke Disease”:ti,ab OR (Disease:ti,ab AND “von Gierke”:ti,ab) OR “von Gierkes Disease”:ti,ab OR (Deficiency:ti,ab AND Glucosephosphatase:ti,ab) OR (Deficiencies:ti,ab AND Glucosephosphatase:ti,ab) OR “Glucosephosphatase Deficienc*”:ti,ab) AND (“Next-generation sequencing”:ti,ab OR NGS:ti,ab OR “Whole exome sequencing”:ti,ab OR WES:ti,ab OR (Sequencing:ti,ab AND Exome:ti,ab) OR “Whole Transcriptome Sequencing”:ti,ab OR (Sequencing:ti,ab AND “Whole Transcriptome”:ti,ab) OR (“Transcriptome Sequencing”:ti,ab AND Whole:ti,ab) OR “Complete Transcriptome Sequencing”:ti,ab OR (Sequencing:ti,ab AND “Complete Transcriptome”:ti,ab) OR (“Transcriptome Sequencing*”:ti,ab AND Complete:ti,ab) OR “Whole Exome Sequencing”:ti,ab OR (“Exome Sequencing”:ti,ab AND Whole:ti,ab) OR (Sequencing:ti,ab AND “Whole Exome”:ti,ab) OR “Complete Exome Sequencing*”:ti,ab OR (“Exome Sequencing*”:ti,ab AND Complete:ti,ab) OR (Sequencing:ti,ab AND “Complete Exome”:ti,ab) OR variants:ti,ab OR “variants types”:ti,ab OR “variants Abundance”:ti,ab OR “variants frequency”:ti,ab OR Variation:ti,ab OR Mutation*:ti,ab)
Scopus
TITLE-ABS-KEY(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR (Disease AND Gierke’s) OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR (Deficienc* AND “Glucose-6-Phosphatase”) OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR (Disease AND “von Gierke”) OR “von Gierke’s Disease” OR (Disease AND “von Gierke’s”) OR “von Gierkes Disease” OR (Deficiency AND Glucosephosphatase) OR (Deficiencies AND Glucosephosphatase) OR “Glucosephosphatase Deficienc*”) AND TITLE-ABS-KEY(“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR (Sequencing AND Exome) OR “Whole Transcriptome Sequencing” OR (Sequencing AND “Whole Transcriptome”) OR (“Transcriptome Sequencing” AND Whole) OR “Complete Transcriptome Sequencing” OR (Sequencing AND “Complete Transcriptome”) OR (“Transcriptome Sequencing*” AND Complete) OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR (Sequencing AND “Whole Exome”) OR “Complete Exome Sequencing*” OR (“Exome Sequencing*” AND Complete) OR (Sequencing AND “Complete Exome”) OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
Web of Science
TS=(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR (Disease AND Gierke’s) OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR (Deficienc* AND “Glucose-6-Phosphatase”) OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR (Disease AND “von Gierke”) OR “von Gierke’s Disease” OR (Disease AND “von Gierke’s”) OR “von Gierkes Disease” OR (Deficiency AND Glucosephosphatase) OR (Deficiencies AND Glucosephosphatase) OR “Glucosephosphatase Deficienc*”) AND TS=(“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR (Sequencing AND Exome) OR “Whole Transcriptome Sequencing” OR (Sequencing AND “Whole Transcriptome”) OR (“Transcriptome Sequencing” AND Whole) OR “Complete Transcriptome Sequencing” OR (Sequencing AND “Complete Transcriptome”) OR (“Transcriptome Sequencing*” AND Complete) OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR (Sequencing AND “Whole Exome”) OR “Complete Exome Sequencing*” OR (“Exome Sequencing*” AND Complete) OR (Sequencing AND “Complete Exome”) OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
Proquest
TI,AB,SU(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR (Disease AND Gierke’s) OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR (Deficienc* AND “Glucose-6-Phosphatase”) OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR (Disease AND “von Gierke”) OR “von Gierke’s Disease” OR (Disease AND “von Gierke’s”) OR “von Gierkes Disease” OR (Deficiency AND Glucosephosphatase) OR (Deficiencies AND Glucosephosphatase) OR “Glucosephosphatase Deficienc*”) AND TI,AB,SU(“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR (Sequencing AND Exome) OR “Whole Transcriptome Sequencing” OR (Sequencing AND “Whole Transcriptome”) OR (“Transcriptome Sequencing” AND Whole) OR “Complete Transcriptome Sequencing” OR (Sequencing AND “Complete Transcriptome”) OR (“Transcriptome Sequencing*” AND Complete) OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR (Sequencing AND “Whole Exome”) OR “Complete Exome Sequencing*” OR (“Exome Sequencing*” AND Complete) OR (Sequencing AND “Complete Exome”) OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
Google scholar
(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR “von Gierke’s Disease” OR “von Gierkes Disease” OR “Glucosephosphatase Deficienc*”) AND (“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR “Whole Transcriptome Sequencing” OR “Complete Transcriptome Sequencing” OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR “Complete Exome Sequencing*” OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
All records=1706
Pubmed=288
Scopus=580
Web of science=327
Embase=384
Proquest=53
Google scholar=74
4, 5 june 2023
An updated search was conducted on 21 May 2025.
Glycogen storage diseases are a group of rare metabolic disorders characterized by defects in glycogen metabolism, leading to glycogen accumulation in tissues, particularly the liver and muscles [1-3]. Among more than 20 subtypes, glycogen storage disease type I (GSD I), or von Gierke disease, is the most common [4]. It results from deficiencies in the glucose-6-phosphatase complex (G6PC1), which plays a critical role in the final step of both glycogenolysis and gluconeogenesis by catalyzing the conversion of glucose-6-phosphate to free glucose. This reaction is essential for maintaining normal blood glucose levels during fasting. Deficiency of this complex prevents the release of glucose into the circulation, leading to severe fasting hypoglycemia and intracellular accumulation of glycogen and lipids, particularly in hepatocytes and renal tubular cells. Based on the affected component of the G6Pase complex, GSD I is classified into two major subtypes: GSD Ia, due to G6PC1 deficiency, and GSD Ib, caused by glucose-6-phosphate transporter (SLC37A4) deficiency. GSD I follows an autosomal recessive inheritance pattern and is more prevalent in populations with high rates of consanguinity [3]. Clinically, GSD I presents in infancy with hypoglycemia, lactic acidemia, hyperlipidemia, hyperuricemia, and hepatomegaly. Chronic metabolic imbalance may lead to long-term complications, including growth retardation, hepatic adenomas with a potential risk of malignant transformation, progressive renal dysfunction, osteoporosis, and delayed puberty. GSD Ib also features neutropenia and inflammatory bowel disease, which predispose affected individuals to recurrent bacterial infections, oral ulcers, and inflammatory bowel disease, thereby contributing to increased morbidity and reduced quality of life [1, 3, 5-7].
Accurate and early diagnosis is essential to prevent complications. While clinical features and biochemical tests are informative, overlapping symptoms with other GSD types, such as GSD III, can complicate diagnosis. Thus, molecular genetic testing, particularly gene mutation analysis, provides a reliable, noninvasive diagnostic tool [5, 8]. Numerous mutations in G6PC1 and SLC37A4 have been reported across various ethnicities. However, the mutation spectrum and frequencies can differ significantly by region [3]. Despite this genetic diversity, the overall incidence of GSD I does not appear to differ significantly among populations [3].
Although numerous mutations of G6PC1 and SLC37A4 have been reported in various ethnic groups, there is limited systematic data on their prevalence and regional patterns in Asian populations, which hinders optimal genetic counseling and the development of effective diagnostic strategies. This study aims to systematically review reported variants of G6PC1 and SLC37A4 in Asian patients with GSD I. Specifically, we sought to identify prevalent mutations in different Asian regions (participants), compare mutation distributions across regions (comparisons), summarize variant frequencies and patterns (outcomes), and synthesize data from observational studies including case reports, case series, and cohort studies (study design). Identifying prevalent mutations and regional patterns can enhance genetic counseling and streamline diagnostic approaches. These findings may also improve patient outcomes through targeted treatment strategies.
Materials and Methods
Information sources and search strategy
This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and meta-analyses (PRISMA) guidelines. A detailed review protocol was developed a priori and prospectively registered in the international prospective register of systematic reviews (PROSPERO) under the registration number CRD42023475081. The registered protocol is publicly available in the PROSPERO database [1]. A comprehensive literature search was conducted in the following electronic databases: Web of Science, PubMed/MEDLINE, Embase, Scopus, and ProQuest, covering studies published from January 1995 to June 4, 2023. The final search was performed on June 4, 2023. In addition, Google Scholar was searched to identify potentially relevant gray literature.
Reference lists of all included articles and relevant reviews were manually screened to identify additional eligible studies. No direct contact with study authors was undertaken to obtain unpublished data. The full electronic search strategies for all databases, including search terms, Boolean operators, and applied limits (language), are provided in Supplementary File S1 to ensure search reproducibility.
Eligibility criteria
Eligible studies were selected based on the PICOS framework.
Participants (P): Asian patients with a confirmed diagnosis of GSD Ia or GSD Ib.
Intervention/Exposure (I): Genetic analysis of G6PC1 and/or SLC37A4 variants.
Comparison (C): Comparison of variant distribution across different Asian regions and populations, where applicable.
Outcomes (O): Type, frequency, and distribution of pathogenic or likely pathogenic variants.
Study design (S): Observational studies, including case reports, case series, cross-sectional, case–control, and cohort studies.
Studies published between 1995 and 2023 in English and available as full-text peer-reviewed articles were included. Conference abstracts, reviews, editorials, non-English publications, and duplicate reports were excluded to ensure data completeness and methodological quality.
Study selection
All records identified through database searching were imported into reference management software, and duplicate records were removed. Two reviewers independently screened the titles and abstracts of all retrieved studies to identify potentially eligible articles. Full texts of the selected studies were then assessed for eligibility based on the predefined inclusion and exclusion criteria. Any disagreements between reviewers at any stage were resolved through discussion or consultation with a third reviewer. Studies meeting the eligibility criteria were included in the final systematic review. As no meta-analysis was conducted, all eligible studies were synthesized narratively.
Data collection process
Data were independently extracted by two reviewers using a predefined data extraction form. Extracted information included publication year, country, sample size, disease subtype, gene variant details, and consanguinity. Any discrepancies between reviewers were resolved through discussion or consultation with a third reviewer. For studies with missing or unclear information, attempts were made to clarify data from the original reports. This process ensured consistency, accuracy, and reliability of the extracted data.
Data items
The following variables were extracted from each included study:
• Study characteristics: authors, publication year, and country;
• Participants: number of patients, disease subtype (GSD Ia or Ib), consanguinity;
• Gene variants: gene name, nucleotide and protein changes, exon location, type of mutation (missense, nonsense, frameshift, splice-site, deletion).
PICOS elements:
• Participants: Asian patients with GSD I;
• Intervention: molecular genetic testing (gene mutation analysis);
• Comparison: variant distribution across regions;
• Outcome: variant frequencies and patterns;
• Study design: observational studies including case reports, case series, and cohort studies;
Only pathogenic or likely pathogenic variants were included; benign variants and polymorphisms were excluded. No additional assumptions or simplifications were applied beyond the predefined criteria.
Data extraction and regional classification
Two reviewers independently screened and extracted data, with discrepancies resolved by a third reviewer. Extracted data included publication year, country, number of patients, disease subtype, mutation details, and consanguinity. To assess geographic variation, studies were grouped into three Asian regions: East Asia, South Asia, and West Asia, following standard United Nations regional definitions.
Only pathogenic or likely pathogenic mutations were included; benign variants and polymorphisms were excluded from the analysis. Pathogenicity classification was performed based on the American College of Medical Genetics and Genomics (ACMG) guidelines, with ClinVar and VarSome used as reference tools to support and validate variant interpretation. In this systematic review, only variants classified as pathogenic or likely pathogenic were included, while benign variants and polymorphisms were excluded.
Risk of bias within studies
Given the descriptive and genetic nature of the included studies—most of which consisted of case reports, case series, cross-sectional studies, and observational cohorts without comparative interventions or outcome measures—a formal quantitative risk-of-bias assessment was considered of limited applicability. Therefore, study quality was evaluated qualitatively at the study level by assessing the clarity of GSD I diagnosis, confirmation of molecular genetic testing, completeness of variant reporting, and transparency of methodological descriptions. Only studies reporting pathogenic or likely pathogenic variants were included to enhance data reliability. These quality considerations were considered during the narrative synthesis and interpretation of variant distribution and frequency patterns.
Risk of bias across studies
As no meta-analysis was performed, statistical assessments of bias across studies, such as publication bias or heterogeneity measures, were not applicable. Nevertheless, potential sources of bias across studies were considered qualitatively, including uneven geographic representation of Asian countries, small sample sizes in some regions, and selective reporting of variants in individual studies. These limitations are acknowledged and discussed in the Discussion section to provide context for interpreting the overall findings.
Data synthesis and summary measures
As this systematic review did not perform a meta-analysis, results were synthesized narratively and using descriptive statistics. Data from individual studies were extracted and grouped by gene (G6PC1 or SLC37A4), country, and Asian region. Principal summary measures included the number of patients with each variant, allele frequencies, and percentage ranges. Frequencies were presented in tables and summarized narratively to compare variant distributions across Asian regions, highlighting prevalent mutations and geographic differences. This descriptive approach ensures clarity and allows assessment of regional patterns and prevalence without statistical pooling or use of consistency measures, such as I².
Given the descriptive nature of the included studies and the focus on genetic variant reporting, no effect estimates, confidence intervals, or meta-analytic measures were calculated. Study-level variant data are presented in detail in tables to allow assessment of variant frequencies and regional patterns.
Results
Search results and study selection
A total of 1,706 records were identified across the Web of Science, PubMed, Embase, Scopus, and ProQuest databases. After removing duplicates, 853 unique records remained. Screening of titles and abstracts excluded 641 studies that did not meet the inclusion criteria. The full texts of 211 articles were subsequently assessed for eligibility, resulting in the exclusion of 140 studies that did not focus on Asian populations, one study lacking a full text, and 29 studies for other reasons (e.g. non-human studies or insufficient data). An updated search and manual review added two additional studies, yielding a total of 70 studies included in this systematic review.
These included studies, published between 1995 and 2023, encompassed 680 patients from 14 countries and represented diverse ethnic groups and physiographic regions across Asia. The characteristics of the included studies, including country, patient numbers, disease subtypes, and mutation details, are summarized in Tables 1 [5-7, 9-11, 22, 26-58], 2 [3, 13-17, 60-65], 3 [63, 67-71], categorized by region and ethnicity.
A PRISMA flow diagram (Figure 1) illustrates the detailed study selection process, including screening, eligibility assessment, and reasons for exclusion at each stage.
Study characteristics
The included studies comprised a total of 70 publications, including case reports, case series, cross-sectional studies, and cohort studies, published between 1995 and 2023. Sample sizes varied widely, ranging from single-patient case reports to multicenter studies involving more than 100 patients. Overall, the studies included 680 patients diagnosed with glycogen storage disease type I, encompassing both GSD Ia (G6PC1-related) and GSD Ib (SLC37A4-related) subtypes.
Participants were patients of Asian origin from 14 countries across East Asia, West Asia, and South Asia. The primary outcomes assessed in all studies were the identification and frequency of pathogenic or likely pathogenic variants in the G6PC1 and/or SLC37A4 genes. Most studies employed molecular genetic techniques such as Sanger sequencing, next-generation sequencing, or whole-exome sequencing for variant detection. Funding sources and risk-of-bias assessments were not consistently reported across studies and were therefore not analyzed at the individual study level.
Due to the descriptive and genetic nature of the included studies, no interventions, comparators, or follow-up periods were applicable. Detailed characteristics of each study, including country, study design, sample size, disease subtype, and identified variants, are summarized in Tables 1 [5-7, 9-11, 22, 26-58], 2 [3, 13-17, 60-65], 3 [63, 67-73].
Across the 70 included studies, qualitative assessment indicated that most studies employed validated molecular genetic methods for variant detection. However, smaller case reports or series from underrepresented regions may have introduced selection bias, and some studies lacked complete details on consanguinity or patient characteristics. These considerations were accounted for in the interpretation of variant distribution and frequency patterns.
Distribution and relative frequencies of G6PC and SLC37A4 gene variants
Tables 4, 5, and 6 present a comprehensive summary of the various G6PC gene variants and their distribution across different regions of Asia, while Tables 7, 8, and 9 present SLC37A4 gene variants.
These tables detail the frequency of each variant type by country and ethnic group, based on individual study data. The total variant frequency for each gene was calculated by dividing the number of occurrences of each variant by the total number of reported variants associated with G6PC or SLC37A4. The column titled “variant frequency related to each study (%)” incorporates data from Tables 1, 2, and 3 for each respective country or ethnicity. The reported values reflect a range of variant frequencies observed in various populations, excluding those derived from case reports.
East Asia
East Asia includes China (with Hong Kong, Macau, and Tibet), Japan, Mongolia, North Korea, South Korea, and Taiwan. From 41 studies, 860 alleles were analyzed, identifying 571 mutated alleles and 83 unique mutations. Of these, 33 unique mutations were associated with the SLC37A4 gene and 50 with the G6PC gene, yielding an overall mutation detection rate of 66.39% (Table 1).
G6Pase gene variants in East Asia
A total of 27 studies across China, Japan, Taiwan, Korea, and Hong Kong investigated G6PC mutations in 245 patients. In total, 50 different mutations were found among 512 mutant alleles. The majority were missense (45), followed by nonsense (9), splice site (7), frameshift (5), and other types. Mutations clustered mostly in exons 2, 3, 4, and 5 (Table 1).
The most prevalent mutation was c.648G>T, representing 67.96% of mutant alleles, and it was widely observed in Japan (153 alleles), Korea (102), China (68), Taiwan (17), and Hong Kong (5). This was followed by c.327G>A (p.R83H) with 5.66% frequency, especially in Chinese and Taiwanese patients. Other notable variants included c.248G>A, c.327G>T, and c.588T>G (p.R170X), each ranging from approximately 1.7% to 2.7% (Table 4).
SLC37A4 gene variants in East Asia
Twelve studies documented 33 SLC37A4 mutations across 59 alleles. The most common were in exons 4, 3, 5, and 10. A splice-site mutation in intron 1 (IVS1 -2 A>C) was also identified. The most frequent variant in China was c.572C>T (p.P191L) (13.55%), while c.521C>T (p.W118R) dominated in Japan (8.62%).
Another significant mutation, c.1094delGCTG/insTC (p.Ser366Argfs*3), produced a truncated protein and accounted for 6.77% of mutations. c.1042_1043delCT, a frameshift mutation in exon 8, was particularly common in China and Korea (5.08%).
Other recurrent variants included c.352T>C, g.1689C>T, c.679delCCTA, and c.935_936delCTG, all found at low to moderate frequencies across regional studies (Tables 1 and 3).
West Asia
Thirteen studies from Turkey, Iran, and Israel reported data on 190 patients, including 118 with GSD Ia and 28 with GSD Ib. Across these, 33 distinct variants were identified in 145 patients (Table 2).
G6PC gene variants in West Asia
Research on G6PC gene variants in West Asia has been limited to three countries: Turkey, Iran, and Israel, despite the region comprising 20 countries, including Armenia, Azerbaijan, and Saudi Arabia.
Nine studies focused on G6PC mutations, reporting 24 distinct variants across 236 alleles, of which 161 were mutated (mutation rate: 68.22%). Israel showed the highest mutation detection rate (100%), followed by Turkey (92.1%) and Iran (16.21%). The most common mutation was c.247C>T (p.R83C), observed in 72.67% of cases and across all three countries. p.W77R (c.229T>C) was the second most frequent (7.45%), especially in Turkish cohorts. c.888G>T (p.G270V) ranked third (6.21%) and was also reported from both Turkey and Israel. Additional low-frequency variants included c.576T>G, c.809G>T, and several private or rare substitutions (Table 5).
SLC37A4 gene variants in West Asia
The mutation spectrum of the SLC37A4 gene has been documented in only two West Asian countries: Turkey and Iran. Six studies from Turkey and Iran reported 9 unique SLC37A4 variants in 27 patients. The overall mutation detection rate was 50%. Most patients (92.6%) came from consanguineous families.
The most frequent variant was c.1042_1043delCT (p.L348Vfs*53), found in 41.66% of cases, with 50–100% frequency across studies. In Iran, notable variants included g.118895235_118901del (946del) (16.66%) and c.1245G>A (p.W415T) (8.33%). In Turkey, c.381+1G>C and c.1043_1044delCT (p.P348Rfs*5) were found in smaller proportions (8.33% each) (Table 8).
South Asia
South Asia encompasses Afghanistan, Bangladesh, Bhutan, India, Maldives, Nepal, Pakistan, and Sri Lanka. Thirteen studies involving 105 patients identified 39 distinct variants across 120 alleles—29 related to GSD Ia and 10 to GSD Ib. Consanguinity was reported in only 15 families (Table 3).
G6PC gene variants in South Asia
Between 2001 and 2022, 29 G6PC mutations were identified in Malaysia, Thailand, India, Pakistan, and Vietnam. India contributed the most data (57 patients, 19 variants). Malaysia reported 42 mutated alleles in 30 patients. Shared mutations included c.648G>T and c.248G>A in Malaysia and Thailand.
The most frequent mutation was c.648G>T (p.L216=), especially in Thailand and Malaysia (23.91%). In India, c.150_151delGT (p.W50Cfs*10) was most common (10.86%), followed by c.355C>G (p.H119D) (6.52%) and c.-225C>G (4.34%). c.248G>A was the third most common overall (10.86%). Mutations mainly clustered in exons 2, 3, and 5 (Table 6).
SLC37A4 gene variants in South Asia
From 2000 to 2022, five studies (14 patients) identified 10 SLC37A4 mutations across India, Pakistan, and Sri Lanka. A total of 28 alleles were analyzed with a 100% detection rate. Four patients were from consanguineous families.
Variants included 4 missense mutations, 2 frameshifts, 4 deletions, and 1 in-frame deletion. Most mutations were located in exons 1 and 2. The most common variant was c.796_797del (p.M266Efs*59) in Pakistan (28.57%), followed by c.898C>T (p.R300C) in India (21.42%). Other less frequent variants included c.169_175del (p.S57Lfs*16) and c.150G>A (p.G50E) (Table 9).
Discussion
This systematic review analyzed 70 studies, including 680 patients from 14 Asian countries, examining mutations in the G6PC1 and SLC37A4 genes among patients with GSD Ia and Ib. By organizing variants geographically across East, West, and South Asia, we identified both shared and region-specific mutation patterns. Key prevalent mutations included c.648G>T in East and South Asia, R83C and p.W77R in West Asia, and c.572C>T in China, providing strong evidence for regionally tailored genetic screening strategies.
These findings have clear clinical relevance: they guide healthcare providers and genetic counselors in accurate diagnosis, targeted molecular testing, and informed counseling, particularly in populations with high consanguinity and founder effects. The data from East Asia are particularly robust, while evidence from other regions remains limited, highlighting the need for further research in underrepresented populations. Overall, this review establishes a comprehensive genetic landscape of GSD I in Asia, supporting both clinical decision-making and future investigations into variant prevalence, regional distribution, and population-specific founder effects.
East Asia showed the highest mutation reporting, particularly from Japan, Korea, China, and Taiwan. The most frequent G6PC1 variant was c.648G>T (c.727G>T), representing 67.96% of all mutations in the region and reaching up to 92% in Korean patients and 81% in Japanese cohorts [5, 7, 9]. This variant causes a splicing error, resulting in a truncated G6Pase protein with approximately 18% enzymatic activity [5]. It appears to be a founder mutation, especially in Japanese populations. Other frequent East Asian mutations included c.327G>A (R83H) and c.248G>A, both missense mutations within exon 2, accounting for 5.6% and 3.1% of mutations respectively, especially among Chinese and Taiwanese populations [6]. These mutations were less common in Korea and Japan (Table 4).
In SLC37A4, East Asian patients commonly exhibited c.572C>T (P191L) and c.352T>C (W118R) (Table 7) [9, 10]. The former represented 23% of mutations in Chinese studies, while the latter was dominant in Japanese GSD Ib patients (17%) [6]. The c.1042_1043delCT deletion also appeared in 4.6% of alleles across Korean and Chinese patients [11, 12]. West Asia showed dominance of the c.247C>T (R83C) mutation in G6PC1, found in 72.67% of cases in studies from Turkey, Iran, and Israel [13-15]. In Iranian cohorts alone, it was reported in over 80% of patients. The p.W77R mutation followed with 11.3% frequency in Turkish patients, while c.888G>T (G270V) made up 6.2% of reported variants [16, 17].
For SLC37A4, the c.1042_1043delCT deletion was the most frequent in Iranian GSD Ib patients (64%), followed by g.118895235_118901del (10.5%) and c.1245G>A (W415T) (7.9%) [18-20].
South Asia displayed a broader mutation spectrum. The c.648G>T mutation appeared again, particularly in Malaysia (31%) and Thailand (27%). In India, the most frequent G6PC1 mutation was c.150_151delGT (W50Cfs*10), comprising 39.5% of cases [21]. Other regionally shared variants were c.248G>A (13%) and c.355C>G (H119D, 8.5%).
In SLC37A4, Pakistani and Indian patients showed frequent c.796_797del (M266Efs*59) and c.898C>T (R300C) mutations, accounting for 44.6% and 28.7% respectively in localized studies [22]. These mutations were rarely identified in other Asian regions.
Comparative data from Europe indicates that R83C, the dominant West Asian variant, is also prevalent in Mediterranean populations but nearly absent in East Asia. Conversely, c.648G>T, common in East and South Asia, is largely undetected in European cohorts. The c.327G>A variant, although present in Asia, occurs at significantly lower frequencies in Europe [23-25].
These findings support the design of regionally customized genetic screening panels. Including prevalent variants such as c.648G>T, R83H, and c.572C>T for East/South Asia and R83C, p.W77R for West Asia can significantly improve diagnostic yield. High consanguinity rates and population-specific founder effects further emphasize the value of population-based genetic approaches in GSD I diagnosis and counseling.
Conclusion
This systematic review highlights substantial genetic diversity in G6PC1 and SLC37A4 mutations among Asian patients with GSD I. Distinct mutation patterns across East, West, and South Asia underscore the necessity for region-specific genetic screening and tailored diagnostic strategies. Key prevalent variants, such as c.648G>T in East/South Asia and c.572C>T in China, enhance diagnostic accuracy and can guide targeted patient management.
These findings provide a comprehensive overview of the genetic landscape of GSD I in Asia, supporting clinical decision-making, informed genetic counseling, and public health planning, particularly in populations with high consanguinity or founder effects. Furthermore, the study highlights gaps in data from underrepresented regions and emphasizes the need for future research employing advanced sequencing approaches, larger cohorts, and inclusive population sampling to better characterize variant frequencies and regional patterns.
Overall, this review underscores the critical role of genetic testing in improving patient outcomes and lays a foundation for personalized medicine approaches in GSD I across diverse Asian populations.
Limitations
This study provides valuable insights into GSD I mutations by analyzing genetic variations across different geographic regions in Asia, the largest continent with a population of approximately 4.7 billion. However, several limitations should be acknowledged. At the study and outcome levels, many included studies had small sample sizes and limited representation from certain countries, which may affect the accuracy and generalizability of variant frequency estimates. At the review level, despite conducting a comprehensive systematic search across multiple databases, some studies may have been missed due to language restrictions, incomplete reporting, or limited accessibility, potentially introducing reporting bias. Furthermore, our analysis included only 32 of 48 Asian countries, encompassing 680 individuals, which may not fully capture the continent’s ethnic and geographic diversity. These limitations highlight the need for broader analyses, particularly in underrepresented populations, and suggest that future studies could benefit from using whole-exome sequencing or other high-throughput genetic approaches to provide a more complete picture of genetic variation in GSD I across Asia.
Ethical Considerations
Compliance with ethical guidelines
The study protocol was approved by the Research Ethics Committee of Isfahan University of Medical Sciences, Isfahan, Iran (Code: IR.ARI.MUI.REC.1403.155).
Funding
This work was supported by Isfahan University of Medical Sciences, Isfahan, Iran (Grant No.: 1402262).
Authors contributions
All authors contributed equally to the conception and design of the study, data collection and analysis, interception of the results and drafting of the manuscript. Each author approved the final version of the manuscript for submission.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors acknowledge the support of the Vice Chancellor for Research at Isfahan University of Medical Sciences, Isfahan, iRAN.
References
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. 2021; 372:n71. [DOI:10.1136/bmj.n71] [PMID]
- Beyzaei Z, Geramizadeh B. Molecular diagnosis of glycogen storage disease type I: A review. EXCLI J. 2019; 18:30-46. [PMID]
- Oguz MM, Aykan E, Yilmaz G, Aytekin C, Karaer K, Açoğlu EA. Glycogen storage disease type 1b: An early onset severe phenotype associated with a novel mutation (IVS4) in the glucose 6-phosphate translocase (SLC37A4) gene in a Turkish patient. Genet Couns. 2014; 25(4):389-94. [PMID]
- Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014; 16(11):e1. [DOI:10.1038/gim.2014.128] [PMID]
- Kajihara S, Matsuhashi S, Yamamoto K, Kido K, Tsuji K, Tanae A, et al. Exon redefinition by a point mutation within exon 5 of the glucose-6-phosphatase gene is the major cause of glycogen storage disease type 1a in Japan. Am J Hum Genet. 1995; 57(3):549-55. [DOI:10.1016/0270-9139(95)95224-1] [PMID]
- Takahashi K, Akanuma J, Matsubara Y, Fujii K, Kure S, Suzuki Y, et al. Heterogeneous mutations in the glucose-6-phosphatase gene in Japanese patients with glycogen storage disease type Ia. Am J Med Genet. 2000; 92(2):90-4. [PMID]
- Kim YM, Choi JH, Lee BH, Kim GH, Kim KM, Yoo HW. Predominance of the c. 648G> T G6PC gene mutation and late complications in Korean patients with glycogen storage disease type Ia. Orphanet J Rare Dis. 2020; 15(1):45. [DOI:10.1186/s13023-020-1321-0] [PMID]
- Zheng BX, Lin Q, Li M, Jin Y. Three novel mutations of the G6PC gene identified in Chinese patients with glycogen storage disease type Ia. Eur J Pediatr. 2015; 174(1):59-63. [DOI:10.1007/s00431-014-2354-y] [PMID]
- Kido J, Nakamura K, Matsumoto S, Mitsubuchi H, Ohura T, Shigematsu Y, et al. Current status of hepatic glycogen storage disease in Japan: Clinical manifestations, treatments and long-term outcomes. J Hum Genet. 2013; 58(5):285-92. [DOI:10.1038/jhg.2013.17] [PMID]
- Kojima K, Kure S, Kamada F, Hao K, Ichinohe A, Sato K, et al. Genetic testing of glycogen storage disease type Ib in Japan: Five novel G6PT1 mutations and a rapid detection method for a prevalent mutation W118R. Mol Genet Metab. 2004; 81(4):343-6. [DOI:10.1016/j.ymgme.2003.12.004] [PMID]
- Wang Z, Zhao R, Jia X, Li X, Ma L, Fu H. Three novel SLC37A4 variants in glycogen storage disease type 1b and a literature review. J Int Med Res. 2023; 51(12):3000605231216633.[DOI:10.1177/03000605231216633] [PMID]
- Ying S, Zhihua Z, Yucan Z, Yu J, Qian L, Bixia Z, et al. Molecular Diagnosis of Panel-Based Next-Generation Sequencing Approach and Clinical Symptoms in Patients With Glycogen Storage Disease: A Single Center Retrospective Study. Front Pediatr. 2020; 8:600446. [DOI:10.3389/fped.2020.600446] [PMID]
- Çakar NE, Gezdirici A, Topuz HŞ, Önal H. Novel variants in Turkish patients with glycogen storage disease. Pediatr Int. 2020; 62(10):1145-50. [DOI:10.1111/ped.14286] [PMID]
- Mahmoud SK, Khorrami A, Rafeey M, Ghergherehchi R, Sima MD. Molecular analysis of glycogen storage disease type Ia in Iranian Azeri Turks: Identification of a novel mutation. J Genet. 2017; 96(1):19-23. [DOI:10.1007/s12041-016-0734-y] [PMID]
- Parvari R, Lei KJ, Bashan N, Hershkovitz E, Korman SH, Barash V, et al. Glycogen storage disease type 1a in Israel: Biochemical, clinical, and mutational studies. Am J Med Genet. 1997; 72(3):286-90. [DOI:10.1002/(sici)1096-8628(19971031)72:3%3C286::aid-ajmg6%3E3.0.co;2-p] [PMID]
- Eminoglu TF, Ezgu FS, Hasanoglu A, Tumer L. Rapid screening of 12 common mutations in Turkish GSD 1a patients using electronic DNA microarray. Gene. 2013; 518(2):346-50. [DOI:10.1016/j.gene.2012.12.104] [PMID]
- Terzioglu M, Emre S, Ozen H, Saltik IN, Koçak N, Ciliv G, et al. Glucose-6-phosphatase gene mutations in Turkish patients with glycogen storage disease type Ia. J Inherit Metab Dis. 2001; 24(8):881-2. [DOI:10.1023/A:1013956611607] [PMID]
- Lam CW, Chan KY, Tong SF, Chan BY, Chan YT, Chan YW. A novel missense mutation (P191L) in the glucose-6-phosphate translocase gene identified in a Chinese family with glycogen storage disease 1b. Hum Mutat. 2000; 16(1):94. [DOI:10.1002/1098-1004(200007)16:13.0.CO;2-Q] [PMID]
- Janecke AR, Lindner M, Erdel M, Mayatepek E, Möslinger D, Podskarbi T, et al. Mutation analysis in glycogen storage disease type 1 non-a. Hum Genet. 2000; 107(3):285-9. [DOI:10.1007/s004390000371] [PMID]
- Meimand SE, Azizi G, Yazdani R, Sanadgol N, Rezaei N. Novel mutation of SLC37A4 in a glycogen storage disease type Ib patient with neutropenia, horseshoe kidney, and arteriovenous malformation: A case report. Immunol Res. 2023; 71(1):107-11. [DOI:10.1007/s12026-022-09320-w] [PMID]
- Parvari R, Moses S, Hershkovitz E, Carmi R, Bashan N. Characterization of the mutations in the glucose-6-phosphatase gene in Israeli patients with glycogen storage disease type 1a: R83C in six Jews and a novel V166G mutation in a Muslim Arab. J Inherit Metab Dis. 1995; 18(1):21-7. [DOI:10.1007/BF00711368] [PMID]
- Han SH, Ki CS, Lee JE, Hong YJ, Son BK, Lee KH, Choe YH, et al. A novel mutation (A148V) in the glucose 6-phosphate translocase (SLC37A4) gene in a Korean patient with glycogen storage disease type 1b. J Korean Med Sci. 2005; 20(3):499-501. [DOI:10.3346/jkms.2005.20.3.499] [PMID]
- Seydewitz HH, Matern D. Molecular genetic analysis of 40 patients with glycogen storage disease type Ia: 100% mutation detection rate and 5 novel mutations. Hum Mutat. 2000; 15(1):115-6. [DOI:10.1002/(sici)1098-1004(200001)15:1%3C115::aid-humu23%3E3.0.co;2-w] [PMID]
- Rake JP, ten Berge AM, Visser G, Verlind E, Niezen-Koning KE, Buys CH, et al. Glycogen storage disease type Ia: Recent experience with mutation analysis, a summary of mutations reported in the literature and a newly developed diagnostic flow chart. Eur J Pediatr. 2000; 159(5):322-30. [DOI:10.1007/s004310051281] [PMID]
- Rake JP, ten Berge AM, Verlind E, Visser G, Niezen-Koning KE, Buys CH, et al. Glycogen storage disease type Ia: four novel mutations (175delGG, R170X, G266V and V338F) identified. Mutations in brief no. 220. Online. Hum Mutat. 1999; 13(2):173. [DOI:10.1002/(sici)1098-1004(1999)13:2%3C173::aid-humu19%3E3.0.co;2-e] [PMID]
- Wu S, Guo S, Fu L, Du C, Luo X. Case Report: Glycogen Storage Disease Type Ia in a Chinese Child Treated With Growth Hormone. Front Pediatr. 2022; 10:921323. [DOI:10.3389/fped.2022.921323] [PMID]
- Du C, Li Z, Wei H, Zhang M, Hu M, Zhang C, et al. Clinical analysis and long-term treatment monitoring of 3 patients with glycogen storage disease type Ib. BMC Med Genomics. 2021; 14(1):81. [DOI:10.1186/s12920-021-00936-9] [PMID]
- Wang F, Wang F, Zhou X, Yi Y, Zhao J. A Novel Lipoprotein Lipase Mutation in an Infant With Glycogen Storage Disease Type-Ib and Severe Hypertriglyceridemia. Front Pediatr. 2021; 9:671536. [DOI:10.3389/fped.2021.671536] [PMID]
- Xu Q, Tang H, Duan L, Zuo X, Shi X, Li Y, et al. A novel SLC37A4 missense mutation in GSD-Ib without hepatomegaly causes enhanced leukocytes endoplasmic reticulum stress and apoptosis. Mol Genet Genomic Med. 2021; 9(1):e1568. [DOI:10.1002/mgg3.1568] [PMID]
- Zhang Y, Sun H, Wan N. Mutation analysis of SLC37A4 in a patient with glycogen storage disease-type Ib. J Int Med Res. 2019; 47(12):5996-6003.[DOI:10.1177/0300060519867819] [PMID]
- Miao H, Zhou J, Yang Q, Liang F, Wang D, Ma N, et al. Long-read sequencing identified a causal structural variant in an exome-negative case and enabled preimplantation genetic diagnosis. Hereditas. 2018; 155:32. [DOI:10.1186/s41065-018-0069-1] [PMID]
- Lu Y, Wang L, Li J, Wu B, Wu H, Luo Y, et al. Molecular genetic analysis and phenotypic characteristics of a consanguineous family with glycogen storage disease type Ia. Mol Med Rep. 2016; 14(4):3251-4. [DOI:10.3892/mmr.2016.5617] [PMID]
- Zhu J, Xing Y, Xing X, Ren A, Ye S, He G. A novel type heterozygous mutation in the glucose-6-phosphatase gene in a Chinese patient with glycogen storage disease Ia. Gene. 2012; 511(1):122-4. [DOI:10.1016/j.gene.2012.09.020] [PMID]
- Li DZ, Liao C, Tang XW. Prenatal diagnosis of glycogen storage disease type Ia, presenting a new mutation in the glucose-6-phosphatase gene. Prenat Diagn. 2007; 27(7):685-6. [DOI:10.1002/pd.1764] [PMID]
- Qiu WJ, Gu XF, Ye J, Han LSh, Zhang YF, Liu XQ. Molecular genetic analysis of glycogen storage disease type Ia in 26 Chinese patients. J Inherit Metab Dis. 2003; 26(8):811-2. [DOI:10.1023/b:boli.0000009992.78840.77] [PMID]
- Yuen YP, Cheng WF, Tong SF, Chan YT, Chan YW, Lam CW. Novel missense mutation (Y24H) in the G6PT1 gene causing glycogen storage disease type 1b. Mol Genet Metab. 2002; 77(3):249-51. [DOI:10.1016/S1096-7192(02)00110-5] [PMID]
- Lei KJ, Chen YT, Chen H, Wong LJ, Liu JL, McConkie-Rosell A, et al. Genetic basis of glycogen storage disease type 1a: Prevalent mutations at the glucose-6-phosphatase locus. Am J Hum Genet. 1995; 57(4):766-71. [PMID]
- Chang CY, Chen CH. Unique glucose-6-phosphatase, catalytic subunit mutation in a child with type ia glycogen storage disease in Taiwan. Journal of Medical Sciences. 2014; 34(5):220-3. [DOI: 10.4103/1011-4564.143651]
- Gu LL, Li XH, Han Y, Zhang DH, Gong QM, Zhang XX. A novel homozygous no-stop mutation in G6PC gene from a Chinese patient with glycogen storage disease type Ia. Gene. 2014; 536(2):362-5. [DOI:10.1016/j.gene.2013.11.059] [PMID]
- Hsiao HJ, Chang HH, Hwu WL, Lam CW, Lee NC, Chien YH. Glycogen storage disease type Ib: The first case in Taiwan. Pediatr Neonatol. 2009; 50(3):125-8. [DOI:10.1016/S1875-9572(09)60048-6] [PMID]
- Wong LJ, Hwu WL, Dai P, Chen TJ. Molecular genetics of glycogen-storage disease type 1a in Chinese patients of Taiwan. Mol Genet Metab. 2001; 72(2):175-80. [DOI:10.1006/mgme.2000.3129] [PMID]
- Chiang SC, Lee YM, Chang MH, Wang TR, Ko TM, Hwu WL. Glucose-6-phosphatase gene mutations in Taiwan Chinese patients with glycogen storage disease type Ia. J Hum Genet. 2000; 45(4):197-9. [DOI:10.1007/s100380070026] [PMID]
- Hwu WL, Chuang SC, Tsai LP, Chang MH, Chuang SM, Wang TR. Glucose-6-phosphatase gene G327A mutation is common in Chinese patients with glycogen storage disease type Ia. Hum Mol Genet. 1995; 4(6):1095-6. [DOI:10.1093/hmg/4.6.1095] [PMID]
- Wu MC, Tsai FJ, Le CC, Lin SP, Wu JY. Identification of a novel missense mutation (T16A) in the glucose-6-phosphatase gene in a Taiwan Chinese patient with glycogen storage disease Ia (Von Gierke disease). Hum Mutat. 2000; 15(4):390. [PMID] [DOI:10.1002/(SICI)1098-1004(200004)15:43.0.CO;2-N]
- Wu MC, Tsai FJ, Lee CC, Tsai CH, Wu JY. A novel missense mutation (H119L) identified in a Taiwan Chinese family with glycogen storage disease Ia (Von Gierke disease). Hum Mutat. 2000; 16(5):447. [DOI:10.1002/1098-1004(200011)16:53.0.CO;2-M] [PMID]
- KO T, Liang Z, Lin Y, Hwu W. Homozygous 341delg/101x of gulcose-6-phosphatase(g6pt) gene causes glycogen storage disease type Ia (Von Gierke disease) in a Chinese patient. Hum Muta. 1999; 13(5):41-4. [DOI: 10.1002/(SICI)1098-1004(1999)13:5<414::AID-HUMU17>3.0.CO;2-5Digital Object Identifie]
- Hwu WL, Chiang SC, Huang SF, Chang MH, Wen WH, Wang TR. Hypercalcaemia in glycogen storage disease type Ia: A case with R83H and 341delG mutations. J Inherit Metab Dis. 1999; 22(8):937-8. [DOI:10.1023/A:1005651809892] [PMID]
- Lee WJ, Lee HM, Chi CS, Shu SG, Lin LY, Lin WH. Genetic analysis of the glucose-6-phosphatase mutation of type 1a glycogen storage disease in a Chinese family. Clin Genet. 1996; 50(4):206-11. [DOI:10.1111/j.1399-0004.1996.tb02627.x] [PMID]
- Lam CW, But WM, Shek CC, Tong SF, Chan YS, Choy KW, et al. Glucose-6-phosphatase gene (727G-->T) splicing mutation is prevalent in Hong Kong Chinese patients with glycogen storage disease type 1a. Clin Genet. 1998; 53(3):184-90. [DOI:10.1111/j.1399-0004.1998.tb02674.x] [PMID]
- Ki CS, Han SH, Kim HJ, Lee SG, Kim EJ, Kim JW, et al. Mutation spectrum of the glucose-6-phosphatase gene and its implication in molecular diagnosis of Korean patients with glycogen storage disease type Ia. Clin Genet. 2004; 65(6):487-9. [DOI:10.1111/j.1399-0004.2004.00260.x] [PMID]
- Goto M, Taki T, Sugie H, Miki Y, Kato H, Hayashi Y. A novel mutation in the glucose-6-phosphatase gene in Korean twins with glycogen storage disease type Ia. J Inherit Metab Dis. 2000; 23(8):851-2. [DOI:10.1023/A:1026777106212] [PMID]
- Nakamura T, Ozawa T, Kawasaki T, Nakamura H, Sugimura H. Glucose-6-phosphatase gene mutations in 20 adult Japanese patients with glycogen storage disease type 1a with reference to hepatic tumors. J Gastroenterol Hepatol. 2001; 16(12):1402-8. [DOI:10.1046/j.1440-1746.2001.02645.x] [PMID]
- Akanuma J, Nishigaki T, Fujii K, Matsubara Y, Inui K, Takahashi K, et al. Glycogen storage disease type Ia: Molecular diagnosis of 51 Japanese patients and characterization of splicing mutations by analysis of ectopically transcribed mRNA from lymphoblastoid cells. Am J Med Genet. 2000; 91(2):107-12. [PMID]
- Okubo M, Aoyama Y, Kishimoto M, Shishiba Y, Murase T. Identification of a point mutation (G727T) in the glucose-6-phosphatase gene in Japanese patients with glycogen storage disease type 1a, and carrier screening in healthy volunteers. Clin Genet. 1997; 51(3):179-83. [DOI:10.1111/j.1399-0004.1997.tb02449.x] [PMID]
- Ihara K, Kuromaru R, Hara T. Genomic structure of the human glucose 6-phosphate translocase gene and novel mutations in the gene of a Japanese patient with glycogen storage disease type Ib. Hum Genet. 1998; 103(4):493-6. [DOI:10.1007/s004390050856] [PMID]
- Kure S, Suzuki Y, Matsubara Y, Sakamoto O, Shintaku H, Isshiki G, et al. Molecular analysis of glycogen storage disease type Ib: identification of a prevalent mutation among Japanese patients and assignment of a putative glucose-6-phosphate translocase gene to chromosome 11. Biochem Biophys Res Commun. 1998; 248(2):426-31.[DOI:10.1006/bbrc.1998.8985] [PMID]
- Eghbali M, Abiri M, Talebi S, Noroozi Z, Shakiba M, Rostami P, et al. Genotype-phenotype correlation and description of two novel mutations in Iranian patients with glycogen storage disease 1b (GSD1b). Orphanet J Rare Dis. 2020; 15(1):35. [DOI:10.1186/s13023-019-1266-3] [PMID]
- Parvari R, Isam J, Moses SW. Glycogen storage disease type 1a in three siblings with the G270V mutation. J Inherit Metab Dis. 1999; 22(2):149-54. [DOI:10.1023/A:1005445802822] [PMID]
- Ersoy M, Uyanik B, Gedikbasi A. Evaluation of Glycogen Storage Patients: Report of Twelve Novel Variants and New Clinical Findings in a Turkish Population. Genes (Basel). 2021; 12(12):1987. [DOI:10.3390/genes12121987] [PMID]
- Dorum S, Gorukmez O. A novel mutation in a newborn baby leading to glycogen storage disease type Ia. Balkan J Med Genet. 2018; 21(2):55-7. [DOI:10.2478/bjmg-2018-0018] [PMID]
- Rafiee Komachali S, Siahpoosh Z, Salehi M. Novel mutation in slc37a4 gene found by whole-exome sequencing in type i glycogen storage disease (Von Gierke) patient in Iran. J Genet Disor Genet Rep, 2022; 11(2). [Link]
- Koshy A, Ramaswamy K, Correa M, Rekha S. Glycogen storage disease: report of 17 cases from southern India. Indian J Gastroenterol. 2006; 25(4):182-4. [PMID]
- Kumar TV, Bhat M, Narayanachar SG, Narayan V, Srikanth AK, Anikar S, et al. Molecular and clinical profiling in a large cohort of Asian Indians with glycogen storage disorders. PLoS One. 2022; 17(7):e0270373. [DOI:10.1371/journal.pone.0270373] [PMID]
- Karthi S, Manimaran P, Varalakshmi P, Ganesh R, Kapoor S, Goyal M, et al. Mutational spectrum and identification of five novel mutations in G6PC1 gene from a cohort of Glycogen Storage Disease Type 1a. Gene. 2019; 700:7-16. [DOI:10.1016/j.gene.2019.03.029] [PMID]
- Karthi S. Glucose-6-phosphatase (G6PC1) promoter polymorphism associated with glycogen storage disease type 1a among the Indian population. RSC advances. 2015; 5(80):65297-302. [DOI:10.1039/C5RA10452A]
- Tamhankar PM, Boggula V, Girisha KM, Phadke SR. Profile of patients with Von Gierke disease from India. Indian Pediatr. 2012; 49(3):228-30. [DOI:10.1007/s13312-012-0056-y] [PMID]
- Meaney C, Cranston T, Lee P, Genet S. A common 2 bp deletion mutation in the glucose-6-phosphatase gene in Indian patients with glycogen storage disease type Ia. J Inherit Metab Dis. 2001; 24(4):517-8. [DOI:10.1023/A:1010598109582] [PMID]
- Dissanayake VH, Jayasinghe JD, Thilakaratne V, Jayasekara RW. A novel mutation in SLC37A4 gene in a Sri Lankan boy with glycogen storage disease type Ib associated with very early onset neutropenia. J Mol Genet Med. 2011; 5:262-3. [DOI:10.4172/1747-0862.1000046] [PMID]
- Hoang NH. Identification of p. His119Leu mutation in the G6PC gene of a Vietnamese patient with glycogen storage disease type Ia. Acad J Biol. 2020; 42(2):93-100. [DOI:10.15625/2615-9023/v42n2.14898]
- Kamolsilp M, Okubo M. G6PC mutations in two patients with glycogen storage disease type Ia in Thailand. Acta Paediatr. 2010; 99(2):164. [DOI:10.1111/j.1651-2227.2009.01549.x] [PMID]
- Abd Rahman AA. Novel mutations of the G6PC gene in Malaysians with glycogen storage disease 1a (GSD1a). Malays J Sci. 2021; 34-45. [DOI:10.22452/mjs.vol40no1.3]
- Sanjeev RK, Shetty S, Harith A, Nair BT, Surendran S. Glycogen storage disease 1b: Diagnosis and workup of a novel mutation. J Nepal Paed Soc. 2016; 36(1):85-7. [DOI:10.3126/jnps.v36i1.12404]
Supplementary Material S1
PubMed
PICo
Population= (“Glycogen storage disease type Ia”[tiab] OR “Glycogen storage disease type Ib”[tiab] OR “Von Gierke disease”[tiab] OR “Type I Glycogen storage disease”[tiab] OR “Type Ia Glycogen storage disease”[tiab] OR “Type Ib Glycogen storage disease”[tiab] OR “Glycogen storage disease type 1”[tiab] OR “Glycogen storage disease type 1a”[tiab] OR “Glycogen storage disease type 1b”[tiab] OR “Type 1 Glycogen storage disease”[tiab] OR “Type 1a Glycogen storage disease”[tiab] OR “Type 1b Glycogen storage disease”[tiab] OR “Glycogen storage disorder type I”[tiab] OR “Glycogen storage disorder type Ia”[tiab] OR “Glycogen storage disorder type Ib”[tiab] OR “Glycogen storage disorder type 1”[tiab] OR “Glycogen storage disorder type 1a”[tiab] OR “Glycogen storage disorder type 1b”[tiab] OR “Type I Glycogen storage disorder”[tiab] OR “Type Ia Glycogen storage disorder”[tiab] OR “Type Ib Glycogen storage disorder”[tiab] OR “Type 1 Glycogen storage disorder”[tiab] OR “Type 1a Glycogen storage disorder”[tiab] OR “Type 1b Glycogen storage disorder”[tiab] OR “Gierke Disease”[tiab] OR (Disease[tiab] AND Gierke[tiab]) OR “Gierke’s Disease”[tiab] OR (Disease[tiab] AND Gierke’s[tiab]) OR “Gierkes Disease”[tiab] OR “Glucose-6-Phosphatase Deficiency”[tiab] OR (Deficienc*[tiab] AND “Glucose-6-Phosphatase”[tiab]) OR “Glucose 6 Phosphatase Deficiency”[tiab] OR “Glucose-6-Phosphatase Deficienc*”[tiab] OR “Glycogen Storage Disease 1”[tiab] OR “GSD I”[tiab] OR “Glycogenosis 1”[tiab] OR “Hepatorenal Glycogen Storage Disease”[tiab] OR “von Gierke Disease”[tiab] OR (Disease[tiab] AND “von Gierke”[tiab]) OR “von Gierke’s Disease”[tiab] OR (Disease[tiab] AND “von Gierke’s”[tiab]) OR “von Gierkes Disease”[tiab] OR (Deficiency[tiab] AND Glucosephosphatase[tiab]) OR (Deficiencies[tiab] AND Glucosephosphatase[tiab]) OR “Glucosephosphatase Deficienc*”[tiab])
Phenomenon of interest= (“Next-generation sequencing”[tiab] OR NGS[tiab] OR “Whole exome sequencing”[tiab] OR WES[tiab] OR (Sequencing[tiab] AND Exome[tiab]) OR “Whole Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Whole Transcriptome”[tiab]) OR (“Transcriptome Sequencing”[tiab] AND Whole[tiab]) OR “Complete Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Complete Transcriptome”[tiab]) OR (“Transcriptome Sequencing*”[tiab] AND Complete[tiab]) OR “Whole Exome Sequencing”[tiab] OR (“Exome Sequencing”[tiab] AND Whole[tiab]) OR (Sequencing[tiab] AND “Whole Exome”[tiab]) OR “Complete Exome Sequencing*”[tiab] OR (“Exome Sequencing*”[tiab] AND Complete[tiab]) OR (Sequencing[tiab] AND “Complete Exome”[tiab]) OR variants[tiab] OR “variants types”[tiab] OR “variants Abundance”[tiab] OR “variants frequency”[tiab] OR Variation[tiab] OR Mutation*[tiab])
Context=-
(“Glycogen storage disease type Ia”[tiab] OR “Glycogen storage disease type Ib”[tiab] OR “Von Gierke disease”[tiab] OR “Type I Glycogen storage disease”[tiab] OR “Type Ia Glycogen storage disease”[tiab] OR “Type Ib Glycogen storage disease”[tiab] OR “Glycogen storage disease type 1”[tiab] OR “Glycogen storage disease type 1a”[tiab] OR “Glycogen storage disease type 1b”[tiab] OR “Type 1 Glycogen storage disease”[tiab] OR “Type 1a Glycogen storage disease”[tiab] OR “Type 1b Glycogen storage disease”[tiab] OR “Glycogen storage disorder type I”[tiab] OR “Glycogen storage disorder type Ia”[tiab] OR “Glycogen storage disorder type Ib”[tiab] OR “Glycogen storage disorder type 1”[tiab] OR “Glycogen storage disorder type 1a”[tiab] OR “Glycogen storage disorder type 1b”[tiab] OR “Type I Glycogen storage disorder”[tiab] OR “Type Ia Glycogen storage disorder”[tiab] OR “Type Ib Glycogen storage disorder”[tiab] OR “Type 1 Glycogen storage disorder”[tiab] OR “Type 1a Glycogen storage disorder”[tiab] OR “Type 1b Glycogen storage disorder”[tiab] OR “Gierke Disease”[tiab] OR (Disease[tiab] AND Gierke[tiab]) OR “Gierke’s Disease”[tiab] OR (Disease[tiab] AND Gierke’s[tiab]) OR “Gierkes Disease”[tiab] OR “Glucose-6-Phosphatase Deficiency”[tiab] OR (Deficienc*[tiab] AND “Glucose-6-Phosphatase”[tiab]) OR “Glucose 6 Phosphatase Deficiency”[tiab] OR “Glucose-6-Phosphatase Deficienc*”[tiab] OR “Glycogen Storage Disease 1”[tiab] OR “GSD I”[tiab] OR “Glycogenosis 1”[tiab] OR “Hepatorenal Glycogen Storage Disease”[tiab] OR “von Gierke Disease”[tiab] OR (Disease[tiab] AND “von Gierke”[tiab]) OR “von Gierke’s Disease”[tiab] OR (Disease[tiab] AND “von Gierke’s”[tiab]) OR “von Gierkes Disease”[tiab] OR (Deficiency[tiab] AND Glucosephosphatase[tiab]) OR (Deficiencies[tiab] AND Glucosephosphatase[tiab]) OR “Glucosephosphatase Deficienc*”[tiab]) AND (“Next-generation sequencing”[tiab] OR NGS[tiab] OR “Whole exome sequencing”[tiab] OR WES[tiab] OR (Sequencing[tiab] AND Exome[tiab]) OR “Whole Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Whole Transcriptome”[tiab]) OR (“Transcriptome Sequencing”[tiab] AND Whole[tiab]) OR “Complete Transcriptome Sequencing”[tiab] OR (Sequencing[tiab] AND “Complete Transcriptome”[tiab]) OR (“Transcriptome Sequencing*”[tiab] AND Complete[tiab]) OR “Whole Exome Sequencing”[tiab] OR (“Exome Sequencing”[tiab] AND Whole[tiab]) OR (Sequencing[tiab] AND “Whole Exome”[tiab]) OR “Complete Exome Sequencing*”[tiab] OR (“Exome Sequencing*”[tiab] AND Complete[tiab]) OR (Sequencing[tiab] AND “Complete Exome”[tiab]) OR variants[tiab] OR “variants types”[tiab] OR “variants Abundance”[tiab] OR “variants frequency”[tiab] OR Variation[tiab] OR Mutation*[tiab])
Embase
(“Glycogen storage disease type Ia”:ti,ab OR “Glycogen storage disease type Ib”:ti,ab OR “Von Gierke disease”:ti,ab OR “Type I Glycogen storage disease”:ti,ab OR “Type Ia Glycogen storage disease”:ti,ab OR “Type Ib Glycogen storage disease”:ti,ab OR “Glycogen storage disease type 1”:ti,ab OR “Glycogen storage disease type 1a”:ti,ab OR “Glycogen storage disease type 1b”:ti,ab OR “Type 1 Glycogen storage disease”:ti,ab OR “Type 1a Glycogen storage disease”:ti,ab OR “Type 1b Glycogen storage disease”:ti,ab OR “Glycogen storage disorder type I”:ti,ab OR “Glycogen storage disorder type Ia”:ti,ab OR “Glycogen storage disorder type Ib”:ti,ab OR “Glycogen storage disorder type 1”:ti,ab OR “Glycogen storage disorder type 1a”:ti,ab OR “Glycogen storage disorder type 1b”:ti,ab OR “Type I Glycogen storage disorder”:ti,ab OR “Type Ia Glycogen storage disorder”:ti,ab OR “Type Ib Glycogen storage disorder”:ti,ab OR “Type 1 Glycogen storage disorder”:ti,ab OR “Type 1a Glycogen storage disorder”:ti,ab OR “Type 1b Glycogen storage disorder”:ti,ab OR “Gierke Disease”:ti,ab OR (Disease:ti,ab AND Gierke:ti,ab) OR “Gierkes Disease”:ti,ab OR “Glucose-6-Phosphatase Deficiency”:ti,ab OR (Deficienc*:ti,ab AND “Glucose-6-Phosphatase”:ti,ab) OR “Glucose 6 Phosphatase Deficiency”:ti,ab OR “Glucose-6-Phosphatase Deficienc*”:ti,ab OR “Glycogen Storage Disease 1”:ti,ab OR “GSD I”:ti,ab OR “Glycogenosis 1”:ti,ab OR “Hepatorenal Glycogen Storage Disease”:ti,ab OR “von Gierke Disease”:ti,ab OR (Disease:ti,ab AND “von Gierke”:ti,ab) OR “von Gierkes Disease”:ti,ab OR (Deficiency:ti,ab AND Glucosephosphatase:ti,ab) OR (Deficiencies:ti,ab AND Glucosephosphatase:ti,ab) OR “Glucosephosphatase Deficienc*”:ti,ab) AND (“Next-generation sequencing”:ti,ab OR NGS:ti,ab OR “Whole exome sequencing”:ti,ab OR WES:ti,ab OR (Sequencing:ti,ab AND Exome:ti,ab) OR “Whole Transcriptome Sequencing”:ti,ab OR (Sequencing:ti,ab AND “Whole Transcriptome”:ti,ab) OR (“Transcriptome Sequencing”:ti,ab AND Whole:ti,ab) OR “Complete Transcriptome Sequencing”:ti,ab OR (Sequencing:ti,ab AND “Complete Transcriptome”:ti,ab) OR (“Transcriptome Sequencing*”:ti,ab AND Complete:ti,ab) OR “Whole Exome Sequencing”:ti,ab OR (“Exome Sequencing”:ti,ab AND Whole:ti,ab) OR (Sequencing:ti,ab AND “Whole Exome”:ti,ab) OR “Complete Exome Sequencing*”:ti,ab OR (“Exome Sequencing*”:ti,ab AND Complete:ti,ab) OR (Sequencing:ti,ab AND “Complete Exome”:ti,ab) OR variants:ti,ab OR “variants types”:ti,ab OR “variants Abundance”:ti,ab OR “variants frequency”:ti,ab OR Variation:ti,ab OR Mutation*:ti,ab)
Scopus
TITLE-ABS-KEY(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR (Disease AND Gierke’s) OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR (Deficienc* AND “Glucose-6-Phosphatase”) OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR (Disease AND “von Gierke”) OR “von Gierke’s Disease” OR (Disease AND “von Gierke’s”) OR “von Gierkes Disease” OR (Deficiency AND Glucosephosphatase) OR (Deficiencies AND Glucosephosphatase) OR “Glucosephosphatase Deficienc*”) AND TITLE-ABS-KEY(“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR (Sequencing AND Exome) OR “Whole Transcriptome Sequencing” OR (Sequencing AND “Whole Transcriptome”) OR (“Transcriptome Sequencing” AND Whole) OR “Complete Transcriptome Sequencing” OR (Sequencing AND “Complete Transcriptome”) OR (“Transcriptome Sequencing*” AND Complete) OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR (Sequencing AND “Whole Exome”) OR “Complete Exome Sequencing*” OR (“Exome Sequencing*” AND Complete) OR (Sequencing AND “Complete Exome”) OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
Web of Science
TS=(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR (Disease AND Gierke’s) OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR (Deficienc* AND “Glucose-6-Phosphatase”) OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR (Disease AND “von Gierke”) OR “von Gierke’s Disease” OR (Disease AND “von Gierke’s”) OR “von Gierkes Disease” OR (Deficiency AND Glucosephosphatase) OR (Deficiencies AND Glucosephosphatase) OR “Glucosephosphatase Deficienc*”) AND TS=(“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR (Sequencing AND Exome) OR “Whole Transcriptome Sequencing” OR (Sequencing AND “Whole Transcriptome”) OR (“Transcriptome Sequencing” AND Whole) OR “Complete Transcriptome Sequencing” OR (Sequencing AND “Complete Transcriptome”) OR (“Transcriptome Sequencing*” AND Complete) OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR (Sequencing AND “Whole Exome”) OR “Complete Exome Sequencing*” OR (“Exome Sequencing*” AND Complete) OR (Sequencing AND “Complete Exome”) OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
Proquest
TI,AB,SU(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR (Disease AND Gierke’s) OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR (Deficienc* AND “Glucose-6-Phosphatase”) OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR (Disease AND “von Gierke”) OR “von Gierke’s Disease” OR (Disease AND “von Gierke’s”) OR “von Gierkes Disease” OR (Deficiency AND Glucosephosphatase) OR (Deficiencies AND Glucosephosphatase) OR “Glucosephosphatase Deficienc*”) AND TI,AB,SU(“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR (Sequencing AND Exome) OR “Whole Transcriptome Sequencing” OR (Sequencing AND “Whole Transcriptome”) OR (“Transcriptome Sequencing” AND Whole) OR “Complete Transcriptome Sequencing” OR (Sequencing AND “Complete Transcriptome”) OR (“Transcriptome Sequencing*” AND Complete) OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR (Sequencing AND “Whole Exome”) OR “Complete Exome Sequencing*” OR (“Exome Sequencing*” AND Complete) OR (Sequencing AND “Complete Exome”) OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
Google scholar
(“Glycogen storage disease type Ia” OR “Glycogen storage disease type Ib” OR “Von Gierke disease” OR “Type I Glycogen storage disease” OR “Type Ia Glycogen storage disease” OR “Type Ib Glycogen storage disease” OR “Glycogen storage disease type 1” OR “Glycogen storage disease type 1a” OR “Glycogen storage disease type 1b” OR “Type 1 Glycogen storage disease” OR “Type 1a Glycogen storage disease” OR “Type 1b Glycogen storage disease” OR “Glycogen storage disorder type I” OR “Glycogen storage disorder type Ia” OR “Glycogen storage disorder type Ib” OR “Glycogen storage disorder type 1” OR “Glycogen storage disorder type 1a” OR “Glycogen storage disorder type 1b” OR “Type I Glycogen storage disorder” OR “Type Ia Glycogen storage disorder” OR “Type Ib Glycogen storage disorder” OR “Type 1 Glycogen storage disorder” OR “Type 1a Glycogen storage disorder” OR “Type 1b Glycogen storage disorder” OR “Gierke Disease” OR (Disease AND Gierke) OR “Gierke’s Disease” OR “Gierkes Disease” OR “Glucose-6-Phosphatase Deficiency” OR “Glucose 6 Phosphatase Deficiency” OR “Glucose-6-Phosphatase Deficienc*” OR “Glycogen Storage Disease 1” OR “GSD I” OR “Glycogenosis 1” OR “Hepatorenal Glycogen Storage Disease” OR “von Gierke Disease” OR “von Gierke’s Disease” OR “von Gierkes Disease” OR “Glucosephosphatase Deficienc*”) AND (“Next-generation sequencing” OR NGS OR “Whole exome sequencing” OR WES OR “Whole Transcriptome Sequencing” OR “Complete Transcriptome Sequencing” OR “Whole Exome Sequencing” OR (“Exome Sequencing” AND Whole) OR “Complete Exome Sequencing*” OR variants OR “variants types” OR “variants Abundance” OR “variants frequency” OR Variation OR Mutation*)
All records=1706
Pubmed=288
Scopus=580
Web of science=327
Embase=384
Proquest=53
Google scholar=74
4, 5 june 2023
An updated search was conducted on 21 May 2025.
Type of Study: Systematic Review |
Subject:
Genetics
Received: 2025/08/13 | Accepted: 2026/02/10 | Published: 2026/04/12
Received: 2025/08/13 | Accepted: 2026/02/10 | Published: 2026/04/12
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
