
Mon, Jul 13, 2026


Volume 14, Issue 2 (April 2026)
J. Pediatr. Rev 2026, 14(2): 197-206 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Davari F, Mehdizadeh S, Hoseini-Aghdam M, Dabbaghzadeh A. Immune Dysregulation and Chronic Mucocutaneous Candidiasis in a Child With STAT1 Gain-of-function Mutation: A Case Report and Literature Review. J. Pediatr. Rev 2026; 14 (2) :197-206
URL: http://jpr.mazums.ac.ir/article-1-802-en.html
URL: http://jpr.mazums.ac.ir/article-1-802-en.html



1- Student Research Committee, Mazandaran University of Medical Sciences, Sari, Iran.
2- Pediatric Infectious Diseases Research Center, Communicable Diseases Institute, Mazandaran University of Medical Sciences, Sari, Iran. ,siamakdabbaghzadeh@gmail.com
2- Pediatric Infectious Diseases Research Center, Communicable Diseases Institute, Mazandaran University of Medical Sciences, Sari, Iran. ,
Keywords: Janus kinase (JAK) inhibitors, Child, Chronic mucocutaneous candidiasis (CMC), Immune dysregulation, STAT1 transcription factor
Full-Text [PDF 888 kb]
(125 Downloads)
| Abstract (HTML) (279 Views)
Full-Text: (40 Views)
Introduction
Fungal infections pose a significant clinical concern, particularly in vulnerable populations, such as patients with COVID-19, where they contribute substantially to morbidity and mortality [1]. Opportunistic pathogens, such as Candida spp., normally commensal members of the human microbiota, can cause localized disease in immunocompetent individuals but may lead to invasive candidiasis in immunocompromised hosts. This life-threatening manifestation is most frequently observed in patients with human immunodeficiency viruses (HIV) infection, primary immunoregulatory disorders, or those receiving immunosuppressive therapies, including corticosteroids and chemotherapy [2, 3].
Candida species are the primary causative agents of chronic mucocutaneous candidiasis (CMC), an immunological disorder characterized by recurrent or persistent fungal infections of the skin, mucous membranes, and nails [4, 5]. These chronic infections predominantly occur in individuals with underlying immunodeficiencies. Among the genetic etiologies, gain-of-function (GOF) mutations in STAT1 constitute the principal cause of CMC, although several other genetic defects have also been implicated [6, 7].
The Janus kinase-signal transducer and activator of transcription (JAK–STAT) pathway, which governs cell growth, proliferation, metabolism, differentiation, and apoptosis, relies on STAT1 as a central transcription factor [8]. Upon stimulation of cellsurface receptors, activated JAK induces STAT1 phosphorylation (p-STAT1), initiating downstream signaling cascades [9]. Over the past 15 years, three classes of monogenic STAT1-related disorders have been identified: autosomal dominant GOF mutations, dominant negative loss-of-function (LOF) variants causing Mendelian susceptibility to mycobacterial disease [10, 11], and biallelic LOF mutations leading to severe combined immunodeficiency (SCID) [6, 7].
A defining feature of GOF mutations, beyond classical cytokine signaling pathways, is the prolonged activation of STAT1 proteins [11]. This aberrant activation disrupts immune homeostasis through two principal mechanisms. First, sustained p-STAT1 drives excessive production of STAT1-dependent cytokines (including interferonγ [IFN-γ], IFN-α/β, and interleukin [IL]-27), establishing a positive feedback loop that perpetuates STAT1 hyperactivation [7, 12-14]. Second, these cytokines suppress Th17 cell differentiation, leading to impaired secretion of IL-17A, IL-17F, and IL-22 [15, 16]. The consequent Th17 deficiency represents a key immunological mechanism underlying the heightened susceptibility to CMC observed in affected patients [17].
CMC occurs in most patients with STAT1 GOF mutations, although the clinical spectrum is highly variable. In addition to recurrent infections, affected individuals frequently present with non-infectious complications such as aneurysms, autoimmune or inflammatory disorders, and malignancies [10, 17]. Management of STAT1 GOF disease typically involves a combination of long-term antifungal therapy, immune modulation, and treatment of associated autoimmune manifestations. Topical and systemic triazoles are commonly employed for antifungal prophylaxis and treatment [10]. Despite these therapeutic strategies, comprehensive characterization of the clinical features, natural history, and prognosis of STAT1 GOF patients remains limited. Here, we describe a 10-year-old girl with a confirmed STAT1 GOF mutation who presented with recurrent mucocutaneous infections, including oral thrush and fungal nail involvement, highlighting the clinical challenges associated with this condition.
Case Presentation
A 10-year-old girl born to consanguineous parents presented to BaAli Hospital in Sari City, Iran, with a history of recurrent oral thrush and chronic fungal nail lesions. Her mucocutaneous symptoms first appeared at 3 years of age and recurred approximately monthly. While episodes of oral thrush typically resolved spontaneously within 7-10 days, the onychomycosis lesions persisted without complete remission (Figure 1 shows the patient’s fungal nail involvement).
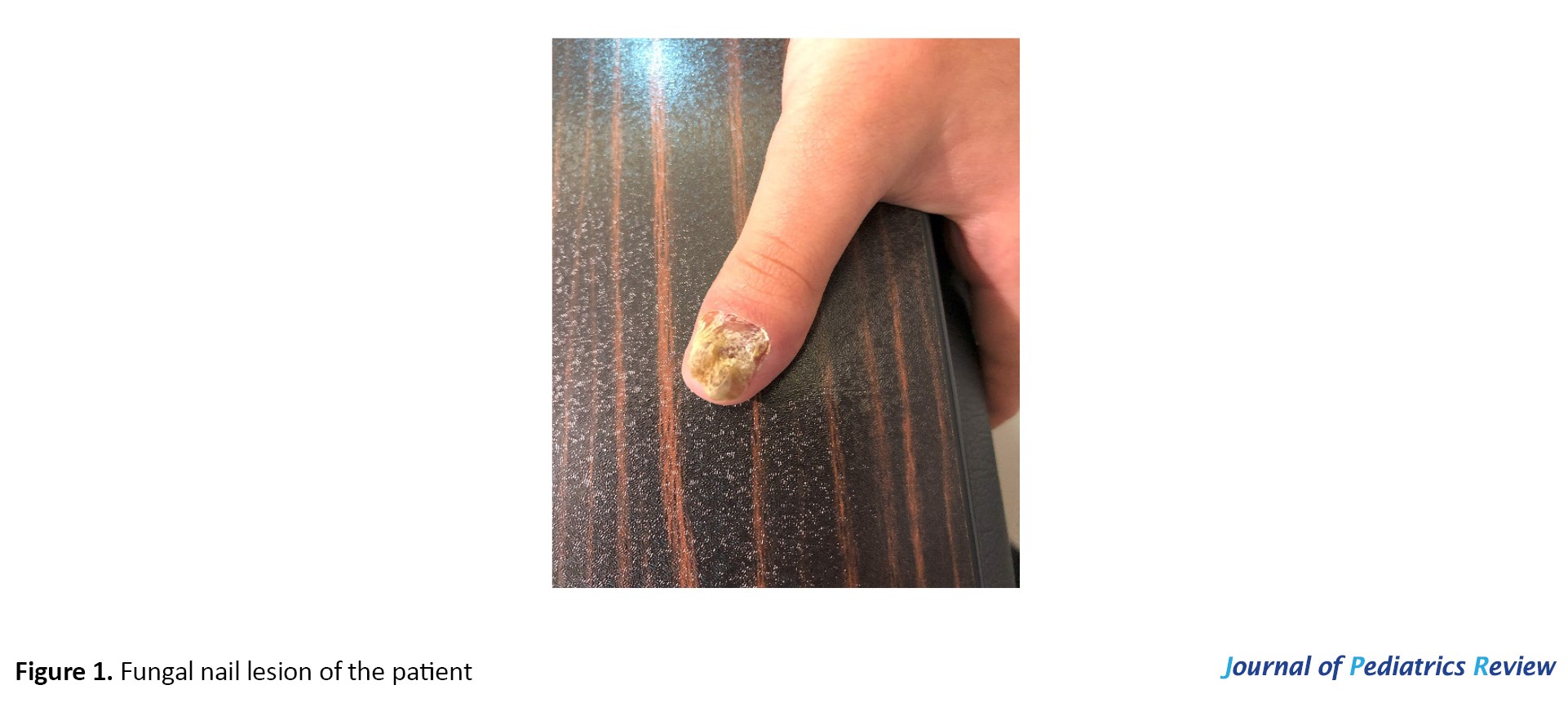 The clinical picture was marked by typical pseudomembranous oral candidiasis occurring about 12 times per year, chronic onychodystrophy involving multiple fingernails, absence of invasive or systemic fungal infection, no concurrent bacterial or viral pathogen susceptibility, and normal growth rate with normal developmental progression.
The clinical picture was marked by typical pseudomembranous oral candidiasis occurring about 12 times per year, chronic onychodystrophy involving multiple fingernails, absence of invasive or systemic fungal infection, no concurrent bacterial or viral pathogen susceptibility, and normal growth rate with normal developmental progression.
The patient’s family history was notable for her father, who required long-term antifungal therapy for recurrent oral thrush and died from esophageal carcinoma at the age of 36. This pattern of familial occurrence strongly suggested an autosomal dominant immunodeficiency. The patient’s favorable response to antifungal therapy was particularly remarkable, as treatment resistance is frequently observed in individuals with STAT1 GOF disorders [17]. This atypical phenotypic presentation prompted a comprehensive immunological evaluation to investigate the underlying cause of immune dysregulation.
Immunological and laboratory results
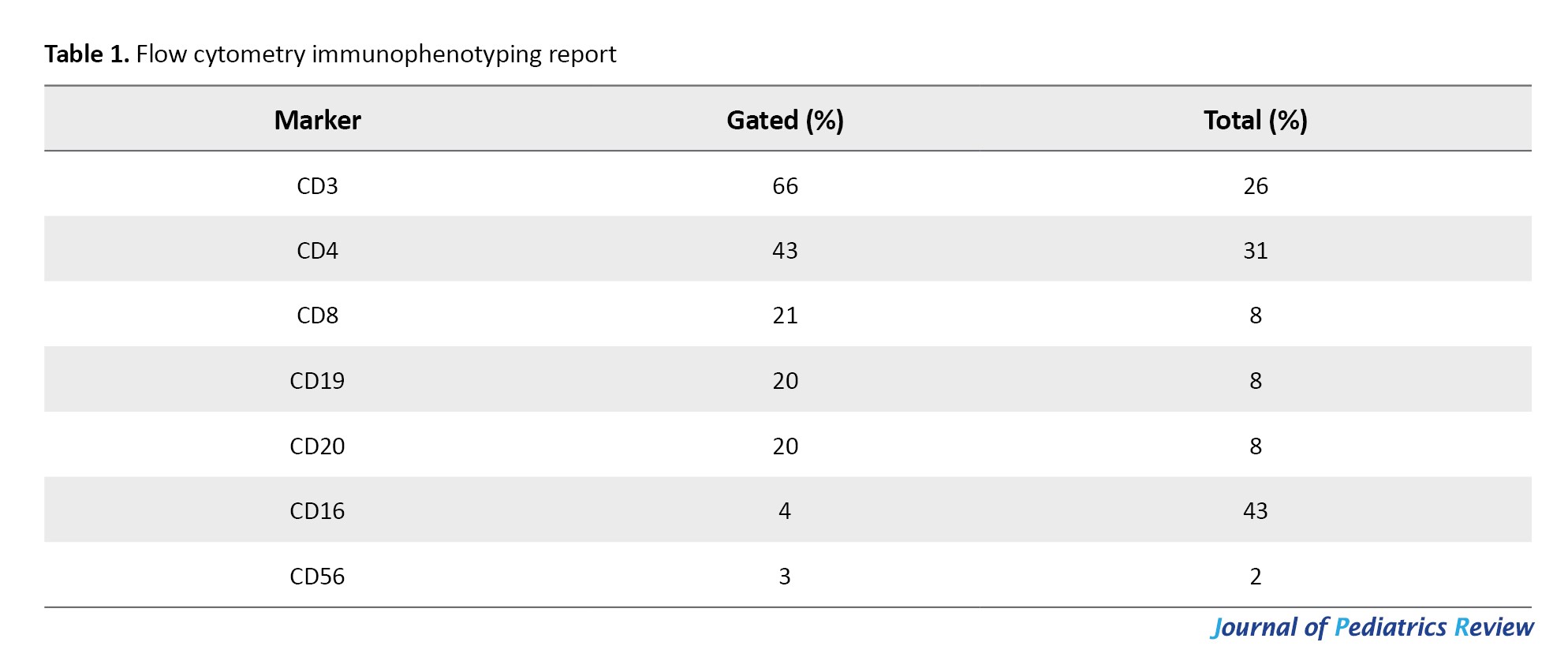
Flow cytometry demonstrated T-cell predominance, with CD3+ cells at 66% (normal: 60–80%), CD4+ at 43%, and CD8+ at 21%, resulting in a CD4/CD8 ratio within normal limits (2.05). B-cell counts (CD19+/CD20+) were within the normal range (20%; normal: 5–20%), and NK-cell counts were low normal (CD56+ 3%) (Table 1).
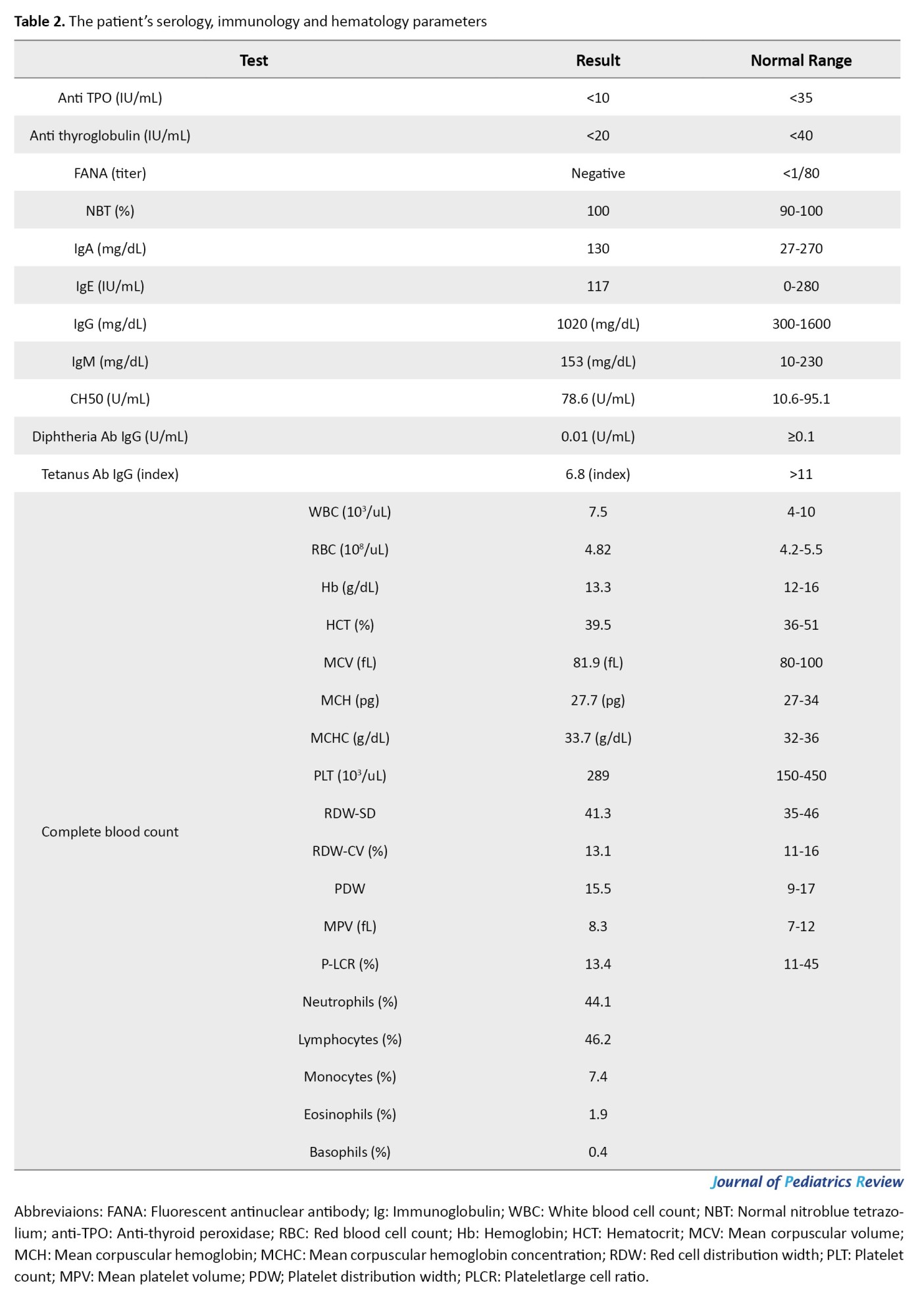
Autoantibody screening was negative for anti-thyroid peroxidase (anti-TPO), anti-thyroglobulin, and fluorescent antinuclear antibody (FANA). Serum immunoglobulin levels were normal (immunoglobulin [Ig]A 130 mg/dL, IgM 153 mg/dL, IgE 117 IU/mL), but the vaccine response was inadequate, with non-protective diphtheria IgG (0.01 IU/mL). Hematologic evaluation revealed a normal white blood cell count (WBC) (7.5×10³/μL), and a normal result on the nitroblue tetrazolium (NBT) test (100%) excluded chronic granulomatous disease. Table 2 summarizes the patient’s hematological parameters.
Genetic analysis
Targeted sequencing identified a heterozygous pathogenic STAT1 mutation (c.C800T, p.A267V; American College of Medical Genetics and Genomics [ACMG] classification: pathogenic), associated with autosomal dominant immunodeficiency 31C (chronic mucocutaneous candidiasis) and immunodeficiency 31A (mycobacterial susceptibility).
Treatment and outcome
The patient was treated with fluconazole prophylaxis (150 mg, every other day) which led to complete resolution of mucocutaneous candidiasis and was combined with baricitinib (2 mg daily), a JAK inhibitor targeting the underlying STAT1 GOF pathology. Supportive therapy included daily sunscreen for prevention of photosensitivity and valacyclovir (500 mg, daily) as antiviral prophylaxis.
Clinical correlations
This case represents an attenuated STAT1 GOF phenotype, characterized by isolated mucocutaneous candidiasis without the typical severe infections or autoimmune manifestations. Remarkably, the patient maintained a normal CD4/CD8 ratio (2.05), in contrast to the lymphocytic dysregulation usually observed in classical STAT1 GOF presentations. Key limitations of the study include the lack of p-STAT1 assays and IL-17/IL-22 profiling, which would provide additional insight into the immune dysregulation underlying this genotype–phenotype association and warrant future investigation.
Discussion
CMC is a rare primary immunodeficiency characterized by recurrent or persistent Candida albicans infections of the skin and mucosal surfaces. Although C. albicans is a commensal organism in healthy individuals, affected patients, primarily children, develop clinically significant infections indicative of impaired antifungal immunity [6].
Genetic susceptibility to CMC has been associated with mutations in several genes, including STAT1, AIRE, CARD9, and Dectin-1 [15]. Among these, STAT1 mutations are the most prevalent and can manifest as either GOF or LOF variants. Over 50% of reported CMC cases are caused by STAT1 GOF mutations, representing the principal molecular mechanism underlying the disease [16].
STAT1 GOF mutations disrupt Th1 and Th17 cell function, resulting in impaired production of critical antifungal cytokines, such as IFN-γ, IL-17, and IL-22, which are essential for mucosal and cutaneous immunity [7]. Additionally, persistent STAT1 activation via IFN-α/β, IFN-γ, and IL-27 signaling further suppresses IL-17 production through mechanisms that are not yet fully understood [12].
While CMC is the most common infectious manifestation in patients with STAT1 GOF mutations, many also experience additional fungal infections and increased susceptibility to bacterial and viral pathogens [10]. These patients frequently present with a spectrum of autoimmune phenomena, including rheumatoid arthritis, autoimmune cytopenias, and immune-dysregulation, polyendocrinopathy, enteropathy, Xlinked (IPEX)-like syndromes, which in some cases may progress to SCID. Respiratory complications, particularly bronchiectasis, have also been reported in a minority of cases [14, 17].
Clinical manifestation
Patients with STAT1 GOF mutations typically present with recurrent mucocutaneous candidiasis involving the oral cavity, genitalia, skin, and nails, with onset usually in early childhood [13]. In the present case, a 10-year-old female exhibited classic CMC manifestations, including oral thrush and onychomycosis, but displayed an unusually restricted phenotype, lacking the bacterial or viral infections and autoimmune manifestations commonly associated with STAT1 GOF mutations. Genetic testing confirmed a pathogenic STAT1 variant (A267V), yet comprehensive serological evaluation—including anti-thyroid peroxidase (anti-TPO), anti-thyroglobulin, and fluorescent antinuclear antibodies (FANA)—revealed no evidence of autoimmunity, in contrast to the typical association of STAT1 GOF with autoimmune disorders [18]. This attenuated phenotype, characterized by isolated mucocutaneous candidiasis without autoimmunity, suggests that the A267V variant may exert hypomorphic effects on STAT1 hyperactivation, partially preserving Th1 and Th17 cell function [19].
A previous report has documented STAT1 GOF mutations associated with CMC and additional cutaneous manifestations, such as rosacea-like demodicosis, underscoring the phenotypic heterogeneity of STAT1 GOF disease [20].
B-cell findings
Patients with STAT1 GOF mutations often exhibit reduced circulating follicular T helper (cTfh) cells, impairing antigen-specific B cell responses [21]. This Tfh deficiency is associated with decreased numbers of memory and naïve B cell, as well as low serum IgG2 (38% of patients) and IgG4 (50%) levels [17]. IL-21 plays a central role in B cell differentiation, antibody synthesis, affinity maturation and isotype switching [22]. However, naïve B cells in STAT1 GOF patients respond poorly to IL-21, resulting in diminished production of IgM, IgG, and IgA [23].
In this case, the patient demonstrated an impaired response to the tetanus-diphtheria (Td) vaccine despite normal B cell counts and baseline immunoglobulin levels. This likely reflects defective T helper cell function, particularly within Th1 and Th17 subsets, which are critical for effective post-vaccination immune activation [24]. Th17 cells are especially important for the development of adaptive immune responses, including robust humoral immunity following vaccination. Excessive p-STAT1 in GOF mutations disrupts cytokine signaling, impairing the differentiation and function of T-helper cells necessary for optimal vaccine responses [25]. For the Td vaccine, a strong Th1 response is essential to generate high-affinity antibodies [26]. Thus, despite competent B cells, dysregulated STAT1 signaling can hinder effective T-helper cell-mediated immunity and vaccine responsiveness.
T cells
Patients with CMC often exhibit T lymphocyte dysfunction, typically characterized by normal T cell counts but impaired responsiveness to Candida antigens [27]. Although not universal, a reduced proportion of Th17 cells in peripheral blood is frequently observed in individuals with STAT1 GOF mutations, contributing to their susceptibility to CMC [7].
Cells from these patients display hyperresponsive STAT1-dependent signaling upon stimulation with type I/II interferons (IFNs) and IL-27. These cytokines, which primarily signal through STAT1, are well-established inhibitors of Th17 cell differentiation in both human and murine systems [7].
STAT1 also influences CD4+ and CD8+ T cell responses; however, hyperactive IFN-γ signaling in GOF mutations promotes excessive Th1 expansion, which can further impair CD8+ T cell differentiation and function, exacerbating CD4+ T cell imbalance [28, 29]. In this case, the CD4/CD8 ratio was within normal limits (2.05), in contrast to the CD4+ predominance typically observed in classical STAT1 GOF presentations [30]. This may reflect compensatory mechanisms specific to the A267V variant or environmental modifiers, such as the microbiome. Alternatively, signaling via other STAT pathways, including STAT3 and STAT5, may help preserve normal T cell subset proportions [31].
Family history implications
Previous studies have demonstrated a strong association between STAT1 GOF mutations and increased susceptibility to esophageal squamous cell carcinoma (SCC) [32]. Notably, the patient’s father died of esophageal cancer at age 36. The proposed mechanism underlying cancer development in STAT1 GOF patients involves chronic inflammation from persistent candidiasis and candidal enzymatic activity, which generates carcinogenic compounds such as nitrosamines [33].
Given this risk, routine monitoring—including careful assessment of gastrointestinal symptoms—is critical for early detection of malignancy [34]. The JAK–STAT pathway inhibitors, particularly JAK inhibitors, such as ruxolitinib and baricitinib, are emerging as therapeutic options to modulate chronic inflammation in STAT1 GOF disease and may help reduce the risk of complications, including SCC and autoimmune manifestations [35, 36]. In the present case, baricitinib was administered to prevent chronic inflammation and its potential sequelae. These considerations underscore the importance of long-term cancer surveillance in patients with STAT1 GOF mutations.
Treatment
The management of STAT1 GOF mutations in patients with CMC typically involves a combination of antifungal, antibacterial, and antiviral therapies, immunomodulatory treatments, such as JAK inhibitors (e.g. ruxolitinib), and management of associated conditions, including autoimmune or autoinflammatory manifestations, with corticosteroids, immunosuppressants, or biologic agents. Patients with STAT1 GOF–associated CMC are particularly prone to recurrent fungal infections, predominantly caused by Candida species, and azoles remain the mainstay of antifungal therapy [15]. In the present case, treatment included fluconazole, the JAK inhibitor baricitinib, and antiviral prophylaxis. Antiviral agents, such as acyclovir and valacyclovir, are commonly employed to prevent herpes simplex virus (HSV) and Varicellazoster virus (VZV) infections, while ganciclovir or valganciclovir was used for CMV prophylaxis. Because no bacterial infections were observed in this patient, antibacterial therapy was not indicated.
Limitations
This study has several limitations. We were unable to assess IL-17 and IL-22 protein expression, and p-STAT1 assays were not performed. In clinical practice, both evaluations are valuable for stratifying patients with CMC according to the underlying immune defect, thereby facilitating more accurate diagnosis, prognosis, and personalized therapy, particularly in cases with overlapping clinical phenotypes. Additionally, no abnormalities were observed in the patient’s total serum IgG level. Although decreased IgG subclass levels (particularly IgG2 and IgG4) have been reported in many patients with CMC and STAT1 GOF mutations [11], these subclasses were not measured in this case. The absence of recurrent pulmonary infections was a key factor in this decision [37]. Nevertheless, assessment of IgG subclasses could provide important diagnostic and management insights for patients with CMC.
The family history of early-onset esophageal cancer underscores the importance of long-term malignancy surveillance in patients with STAT1 GOF, as chronic inflammation and immune dysregulation may increase cancer risk. Although this case expands our understanding of genotype–phenotype correlations in STAT1 GOF disorders, the absence of IL-17 and IL-22 and p-STAT1 testing represents a diagnostic limitation.
Future studies should investigate how specific STAT1 variants modulate Th1/Th17 balance and susceptibility to malignancy to optimize monitoring and therapeutic strategies. Overall, this report emphasizes the need for individualized management in STAT1 GOF syndromes, combining antifungal prophylaxis, targeted immunomodulation, and vigilant cancer screening, while encouraging the development of therapies that restore immune homeostasis without compromising host defense.
Conclusion
This case highlights the broad clinical spectrum of STAT1 GOF mutations, demonstrating that hypomorphic variants, such as A267V, can present with isolated mucocutaneous candidiasis while sparing patients from the severe infections, autoimmunity, and lymphocytic dysregulation typically seen in typical STAT1 GOF syndromes. The the patient’s CD4/CD8 ratio within the normal range, normal immunoglobulin levels, and absence of autoimmunity suggest that the A267V variant may partially maintain immune homeostasis. Nevertheless, antifungal therapy and JAK inhibition with baricitinib were essential for disease control.
Ethical Considerations
Compliance with ethical guidelines
The study was approved by the Ethics Committee of Mazandaran University of Medical Sciences, Sari, Iran (Code IR.MAZUMS.REC.1404.387). Permission was obtained from the child’s parents to publish this case report and any associated photographs. This article adheres to all relevant ethical principles. The patient and her parents have provided written informed consent for publication of this manuscript and collecting images.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
All authors contributed equally to the preparation of this manuscript.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors are grateful to the patient's family for their informed consent. The authors also thank Fatemeh Hosseinzadeh, manager of Pediatric Infectious Diseases Research Center for her support in the preparation of this manuscript.
References
Fungal infections pose a significant clinical concern, particularly in vulnerable populations, such as patients with COVID-19, where they contribute substantially to morbidity and mortality [1]. Opportunistic pathogens, such as Candida spp., normally commensal members of the human microbiota, can cause localized disease in immunocompetent individuals but may lead to invasive candidiasis in immunocompromised hosts. This life-threatening manifestation is most frequently observed in patients with human immunodeficiency viruses (HIV) infection, primary immunoregulatory disorders, or those receiving immunosuppressive therapies, including corticosteroids and chemotherapy [2, 3].
Candida species are the primary causative agents of chronic mucocutaneous candidiasis (CMC), an immunological disorder characterized by recurrent or persistent fungal infections of the skin, mucous membranes, and nails [4, 5]. These chronic infections predominantly occur in individuals with underlying immunodeficiencies. Among the genetic etiologies, gain-of-function (GOF) mutations in STAT1 constitute the principal cause of CMC, although several other genetic defects have also been implicated [6, 7].
The Janus kinase-signal transducer and activator of transcription (JAK–STAT) pathway, which governs cell growth, proliferation, metabolism, differentiation, and apoptosis, relies on STAT1 as a central transcription factor [8]. Upon stimulation of cellsurface receptors, activated JAK induces STAT1 phosphorylation (p-STAT1), initiating downstream signaling cascades [9]. Over the past 15 years, three classes of monogenic STAT1-related disorders have been identified: autosomal dominant GOF mutations, dominant negative loss-of-function (LOF) variants causing Mendelian susceptibility to mycobacterial disease [10, 11], and biallelic LOF mutations leading to severe combined immunodeficiency (SCID) [6, 7].
A defining feature of GOF mutations, beyond classical cytokine signaling pathways, is the prolonged activation of STAT1 proteins [11]. This aberrant activation disrupts immune homeostasis through two principal mechanisms. First, sustained p-STAT1 drives excessive production of STAT1-dependent cytokines (including interferonγ [IFN-γ], IFN-α/β, and interleukin [IL]-27), establishing a positive feedback loop that perpetuates STAT1 hyperactivation [7, 12-14]. Second, these cytokines suppress Th17 cell differentiation, leading to impaired secretion of IL-17A, IL-17F, and IL-22 [15, 16]. The consequent Th17 deficiency represents a key immunological mechanism underlying the heightened susceptibility to CMC observed in affected patients [17].
CMC occurs in most patients with STAT1 GOF mutations, although the clinical spectrum is highly variable. In addition to recurrent infections, affected individuals frequently present with non-infectious complications such as aneurysms, autoimmune or inflammatory disorders, and malignancies [10, 17]. Management of STAT1 GOF disease typically involves a combination of long-term antifungal therapy, immune modulation, and treatment of associated autoimmune manifestations. Topical and systemic triazoles are commonly employed for antifungal prophylaxis and treatment [10]. Despite these therapeutic strategies, comprehensive characterization of the clinical features, natural history, and prognosis of STAT1 GOF patients remains limited. Here, we describe a 10-year-old girl with a confirmed STAT1 GOF mutation who presented with recurrent mucocutaneous infections, including oral thrush and fungal nail involvement, highlighting the clinical challenges associated with this condition.
Case Presentation
A 10-year-old girl born to consanguineous parents presented to BaAli Hospital in Sari City, Iran, with a history of recurrent oral thrush and chronic fungal nail lesions. Her mucocutaneous symptoms first appeared at 3 years of age and recurred approximately monthly. While episodes of oral thrush typically resolved spontaneously within 7-10 days, the onychomycosis lesions persisted without complete remission (Figure 1 shows the patient’s fungal nail involvement).
The patient’s family history was notable for her father, who required long-term antifungal therapy for recurrent oral thrush and died from esophageal carcinoma at the age of 36. This pattern of familial occurrence strongly suggested an autosomal dominant immunodeficiency. The patient’s favorable response to antifungal therapy was particularly remarkable, as treatment resistance is frequently observed in individuals with STAT1 GOF disorders [17]. This atypical phenotypic presentation prompted a comprehensive immunological evaluation to investigate the underlying cause of immune dysregulation.
Immunological and laboratory results
Flow cytometry demonstrated T-cell predominance, with CD3+ cells at 66% (normal: 60–80%), CD4+ at 43%, and CD8+ at 21%, resulting in a CD4/CD8 ratio within normal limits (2.05). B-cell counts (CD19+/CD20+) were within the normal range (20%; normal: 5–20%), and NK-cell counts were low normal (CD56+ 3%) (Table 1).
Autoantibody screening was negative for anti-thyroid peroxidase (anti-TPO), anti-thyroglobulin, and fluorescent antinuclear antibody (FANA). Serum immunoglobulin levels were normal (immunoglobulin [Ig]A 130 mg/dL, IgM 153 mg/dL, IgE 117 IU/mL), but the vaccine response was inadequate, with non-protective diphtheria IgG (0.01 IU/mL). Hematologic evaluation revealed a normal white blood cell count (WBC) (7.5×10³/μL), and a normal result on the nitroblue tetrazolium (NBT) test (100%) excluded chronic granulomatous disease. Table 2 summarizes the patient’s hematological parameters.
Genetic analysis
Targeted sequencing identified a heterozygous pathogenic STAT1 mutation (c.C800T, p.A267V; American College of Medical Genetics and Genomics [ACMG] classification: pathogenic), associated with autosomal dominant immunodeficiency 31C (chronic mucocutaneous candidiasis) and immunodeficiency 31A (mycobacterial susceptibility).
Treatment and outcome
The patient was treated with fluconazole prophylaxis (150 mg, every other day) which led to complete resolution of mucocutaneous candidiasis and was combined with baricitinib (2 mg daily), a JAK inhibitor targeting the underlying STAT1 GOF pathology. Supportive therapy included daily sunscreen for prevention of photosensitivity and valacyclovir (500 mg, daily) as antiviral prophylaxis.
Clinical correlations
This case represents an attenuated STAT1 GOF phenotype, characterized by isolated mucocutaneous candidiasis without the typical severe infections or autoimmune manifestations. Remarkably, the patient maintained a normal CD4/CD8 ratio (2.05), in contrast to the lymphocytic dysregulation usually observed in classical STAT1 GOF presentations. Key limitations of the study include the lack of p-STAT1 assays and IL-17/IL-22 profiling, which would provide additional insight into the immune dysregulation underlying this genotype–phenotype association and warrant future investigation.
Discussion
CMC is a rare primary immunodeficiency characterized by recurrent or persistent Candida albicans infections of the skin and mucosal surfaces. Although C. albicans is a commensal organism in healthy individuals, affected patients, primarily children, develop clinically significant infections indicative of impaired antifungal immunity [6].
Genetic susceptibility to CMC has been associated with mutations in several genes, including STAT1, AIRE, CARD9, and Dectin-1 [15]. Among these, STAT1 mutations are the most prevalent and can manifest as either GOF or LOF variants. Over 50% of reported CMC cases are caused by STAT1 GOF mutations, representing the principal molecular mechanism underlying the disease [16].
STAT1 GOF mutations disrupt Th1 and Th17 cell function, resulting in impaired production of critical antifungal cytokines, such as IFN-γ, IL-17, and IL-22, which are essential for mucosal and cutaneous immunity [7]. Additionally, persistent STAT1 activation via IFN-α/β, IFN-γ, and IL-27 signaling further suppresses IL-17 production through mechanisms that are not yet fully understood [12].
While CMC is the most common infectious manifestation in patients with STAT1 GOF mutations, many also experience additional fungal infections and increased susceptibility to bacterial and viral pathogens [10]. These patients frequently present with a spectrum of autoimmune phenomena, including rheumatoid arthritis, autoimmune cytopenias, and immune-dysregulation, polyendocrinopathy, enteropathy, Xlinked (IPEX)-like syndromes, which in some cases may progress to SCID. Respiratory complications, particularly bronchiectasis, have also been reported in a minority of cases [14, 17].
Clinical manifestation
Patients with STAT1 GOF mutations typically present with recurrent mucocutaneous candidiasis involving the oral cavity, genitalia, skin, and nails, with onset usually in early childhood [13]. In the present case, a 10-year-old female exhibited classic CMC manifestations, including oral thrush and onychomycosis, but displayed an unusually restricted phenotype, lacking the bacterial or viral infections and autoimmune manifestations commonly associated with STAT1 GOF mutations. Genetic testing confirmed a pathogenic STAT1 variant (A267V), yet comprehensive serological evaluation—including anti-thyroid peroxidase (anti-TPO), anti-thyroglobulin, and fluorescent antinuclear antibodies (FANA)—revealed no evidence of autoimmunity, in contrast to the typical association of STAT1 GOF with autoimmune disorders [18]. This attenuated phenotype, characterized by isolated mucocutaneous candidiasis without autoimmunity, suggests that the A267V variant may exert hypomorphic effects on STAT1 hyperactivation, partially preserving Th1 and Th17 cell function [19].
A previous report has documented STAT1 GOF mutations associated with CMC and additional cutaneous manifestations, such as rosacea-like demodicosis, underscoring the phenotypic heterogeneity of STAT1 GOF disease [20].
B-cell findings
Patients with STAT1 GOF mutations often exhibit reduced circulating follicular T helper (cTfh) cells, impairing antigen-specific B cell responses [21]. This Tfh deficiency is associated with decreased numbers of memory and naïve B cell, as well as low serum IgG2 (38% of patients) and IgG4 (50%) levels [17]. IL-21 plays a central role in B cell differentiation, antibody synthesis, affinity maturation and isotype switching [22]. However, naïve B cells in STAT1 GOF patients respond poorly to IL-21, resulting in diminished production of IgM, IgG, and IgA [23].
In this case, the patient demonstrated an impaired response to the tetanus-diphtheria (Td) vaccine despite normal B cell counts and baseline immunoglobulin levels. This likely reflects defective T helper cell function, particularly within Th1 and Th17 subsets, which are critical for effective post-vaccination immune activation [24]. Th17 cells are especially important for the development of adaptive immune responses, including robust humoral immunity following vaccination. Excessive p-STAT1 in GOF mutations disrupts cytokine signaling, impairing the differentiation and function of T-helper cells necessary for optimal vaccine responses [25]. For the Td vaccine, a strong Th1 response is essential to generate high-affinity antibodies [26]. Thus, despite competent B cells, dysregulated STAT1 signaling can hinder effective T-helper cell-mediated immunity and vaccine responsiveness.
T cells
Patients with CMC often exhibit T lymphocyte dysfunction, typically characterized by normal T cell counts but impaired responsiveness to Candida antigens [27]. Although not universal, a reduced proportion of Th17 cells in peripheral blood is frequently observed in individuals with STAT1 GOF mutations, contributing to their susceptibility to CMC [7].
Cells from these patients display hyperresponsive STAT1-dependent signaling upon stimulation with type I/II interferons (IFNs) and IL-27. These cytokines, which primarily signal through STAT1, are well-established inhibitors of Th17 cell differentiation in both human and murine systems [7].
STAT1 also influences CD4+ and CD8+ T cell responses; however, hyperactive IFN-γ signaling in GOF mutations promotes excessive Th1 expansion, which can further impair CD8+ T cell differentiation and function, exacerbating CD4+ T cell imbalance [28, 29]. In this case, the CD4/CD8 ratio was within normal limits (2.05), in contrast to the CD4+ predominance typically observed in classical STAT1 GOF presentations [30]. This may reflect compensatory mechanisms specific to the A267V variant or environmental modifiers, such as the microbiome. Alternatively, signaling via other STAT pathways, including STAT3 and STAT5, may help preserve normal T cell subset proportions [31].
Family history implications
Previous studies have demonstrated a strong association between STAT1 GOF mutations and increased susceptibility to esophageal squamous cell carcinoma (SCC) [32]. Notably, the patient’s father died of esophageal cancer at age 36. The proposed mechanism underlying cancer development in STAT1 GOF patients involves chronic inflammation from persistent candidiasis and candidal enzymatic activity, which generates carcinogenic compounds such as nitrosamines [33].
Given this risk, routine monitoring—including careful assessment of gastrointestinal symptoms—is critical for early detection of malignancy [34]. The JAK–STAT pathway inhibitors, particularly JAK inhibitors, such as ruxolitinib and baricitinib, are emerging as therapeutic options to modulate chronic inflammation in STAT1 GOF disease and may help reduce the risk of complications, including SCC and autoimmune manifestations [35, 36]. In the present case, baricitinib was administered to prevent chronic inflammation and its potential sequelae. These considerations underscore the importance of long-term cancer surveillance in patients with STAT1 GOF mutations.
Treatment
The management of STAT1 GOF mutations in patients with CMC typically involves a combination of antifungal, antibacterial, and antiviral therapies, immunomodulatory treatments, such as JAK inhibitors (e.g. ruxolitinib), and management of associated conditions, including autoimmune or autoinflammatory manifestations, with corticosteroids, immunosuppressants, or biologic agents. Patients with STAT1 GOF–associated CMC are particularly prone to recurrent fungal infections, predominantly caused by Candida species, and azoles remain the mainstay of antifungal therapy [15]. In the present case, treatment included fluconazole, the JAK inhibitor baricitinib, and antiviral prophylaxis. Antiviral agents, such as acyclovir and valacyclovir, are commonly employed to prevent herpes simplex virus (HSV) and Varicellazoster virus (VZV) infections, while ganciclovir or valganciclovir was used for CMV prophylaxis. Because no bacterial infections were observed in this patient, antibacterial therapy was not indicated.
Limitations
This study has several limitations. We were unable to assess IL-17 and IL-22 protein expression, and p-STAT1 assays were not performed. In clinical practice, both evaluations are valuable for stratifying patients with CMC according to the underlying immune defect, thereby facilitating more accurate diagnosis, prognosis, and personalized therapy, particularly in cases with overlapping clinical phenotypes. Additionally, no abnormalities were observed in the patient’s total serum IgG level. Although decreased IgG subclass levels (particularly IgG2 and IgG4) have been reported in many patients with CMC and STAT1 GOF mutations [11], these subclasses were not measured in this case. The absence of recurrent pulmonary infections was a key factor in this decision [37]. Nevertheless, assessment of IgG subclasses could provide important diagnostic and management insights for patients with CMC.
The family history of early-onset esophageal cancer underscores the importance of long-term malignancy surveillance in patients with STAT1 GOF, as chronic inflammation and immune dysregulation may increase cancer risk. Although this case expands our understanding of genotype–phenotype correlations in STAT1 GOF disorders, the absence of IL-17 and IL-22 and p-STAT1 testing represents a diagnostic limitation.
Future studies should investigate how specific STAT1 variants modulate Th1/Th17 balance and susceptibility to malignancy to optimize monitoring and therapeutic strategies. Overall, this report emphasizes the need for individualized management in STAT1 GOF syndromes, combining antifungal prophylaxis, targeted immunomodulation, and vigilant cancer screening, while encouraging the development of therapies that restore immune homeostasis without compromising host defense.
Conclusion
This case highlights the broad clinical spectrum of STAT1 GOF mutations, demonstrating that hypomorphic variants, such as A267V, can present with isolated mucocutaneous candidiasis while sparing patients from the severe infections, autoimmunity, and lymphocytic dysregulation typically seen in typical STAT1 GOF syndromes. The the patient’s CD4/CD8 ratio within the normal range, normal immunoglobulin levels, and absence of autoimmunity suggest that the A267V variant may partially maintain immune homeostasis. Nevertheless, antifungal therapy and JAK inhibition with baricitinib were essential for disease control.
Ethical Considerations
Compliance with ethical guidelines
The study was approved by the Ethics Committee of Mazandaran University of Medical Sciences, Sari, Iran (Code IR.MAZUMS.REC.1404.387). Permission was obtained from the child’s parents to publish this case report and any associated photographs. This article adheres to all relevant ethical principles. The patient and her parents have provided written informed consent for publication of this manuscript and collecting images.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
All authors contributed equally to the preparation of this manuscript.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors are grateful to the patient's family for their informed consent. The authors also thank Fatemeh Hosseinzadeh, manager of Pediatric Infectious Diseases Research Center for her support in the preparation of this manuscript.
References
- Domán M, Bányai K. COVID-19-associated fungal infections: An urgent need for alternative therapeutic approach? Front Microbiol. 2022; 13:919501. [DOI:10.3389/fmicb.2022.919501] [PMID]
- Kaushik N, Pujalte GG, Reese ST. Superficial fungal infections. Prim Care. 2015; 42(4):501-16. [DOI:10.1016/j.pop.2015.08.004] [PMID]
- Reddy GKK, Padmavathi AR, Nancharaiah Y. Fungal infections: Pathogenesis, antifungals and alternate treatment approaches. Curr Res Microb Sci. 2022; 3:100137. [DOI:10.1016/j.crmicr.2022.100137] [PMID]
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international :union: of immunological societies expert committee. J Clin Immunol. 2022; 42(7):1473-507. [DOI:10.1007/s10875-022-01289-3] [PMID]
- Puel A. Human inborn errors of immunity underlying superficial or invasive candidiasis. Human Genetics. 2020; 139(6):1011-22. [DOI:10.1007/s00439-020-02141-7] [PMID]
- van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011; 365(1):54-61. [DOI:10.1056/NEJMoa1100102] [PMID]
- Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011; 208(8):1635-48. [DOI:10.1084/jem.20110958] [PMID]
- O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013; 368(2):161-70. [DOI:10.1056/NEJMra1202117] [PMID]
- Xie Y, Shao F, Lei J, Huang N, Fan Z, Yu H. Case report: A STAT1 gain-of-function mutation causes a syndrome of combined immunodeficiency, autoimmunity and pure red cell aplasia. Front Immunol. 2022; 13:928213. [DOI:10.3389/fimmu.2022.928213] [PMID]
- Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R,et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol. 2016; 36(1):73-84. [DOI:10.1007/s10875-015-0214-9] [PMID]
- Okada S, Asano T, Moriya K, Boisson-Dupuis S, Kobayashi M, Casanova JL, et al. Human STAT1 gain-of-function heterozygous mutations: Chronic mucocutaneous candidiasis and type I interferonopathy. J Clin Immunol. 2020; 40(8):1065-81. [DOI:10.1007/s10875-020-00847-x] [PMID]
- Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012; 24(4):364-78. [DOI:10.1016/j.coi.2012.04.011] [PMID]
- Ouederni M, Sanal O, Ikincioğullari A, Tezcan I, Dogu F, Sologuren I, et al. Clinical features of candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis. 2014; 58(2):204-13. [DOI:10.1093/cid/cit722] [PMID]
- Takeda MR, Bansal M, Kamerman-Kretzmer RJ, Church J, Ji J, Warren M. Bronchiectasis and bronchiolectasis with severe herniating pattern associated with STAT1 Gain-of-function mutation: detailed clinicopathological findings. Pediatr Dev Pathol. 2021; 24(2):131-6. [DOI:10.1177/1093526620985950] [PMID]
- Okada S, Puel A, Casanova JL, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunology. 2016; 5(12):e114. [DOI:10.1038/cti.2016.71] [PMID]
- Wang Z, Zhang Y, Ma W. Chronic mucocutaneous candidiasis: A case report. Clin Cosmet Investig Dermatol. 2023; 16:231-236. [DOI:10.2147/CCID.S396802] [PMID]
- Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016; 127(25):3154-64. [DOI:10.1182/blood-2015-11-679902] [PMID]
- Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010; 207(2):291-7. [DOI:10.1084/jem.20091983] [PMID]
- Antoniadi M, Tzanoudaki M, Karkanis V, Kostaridou S, Tsagris V. STAT1 gain-of-function mutation in chronic mucocutaneous candidiasis. Indian J Pediatr. 2024; 91(12):1312. [DOI:10.1007/s12098-024-05248-1] [PMID]
- Martinot M, Korganow AS, Wald M, Second J, Birckel E, Mahé A, et al. Case report: A new gain-of-function mutation of stat1 identified in a patient with chronic mucocutaneous candidiasis and rosacea-like demodicosis: An emerging association. Front Immunol. 2021; 12:760019. [DOI:10.3389/fimmu.2021.760019] [PMID]
- Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol. 2015; 136(4):993-1006.e1. [PMID]
- Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, et al. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: Predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol. 2007; 179(12):8180-90. [DOI:10.4049/jimmunol.179.12.8180] [PMID]
- Egri N, Esteve-Solé A, Deyà-Martínez À, Ortiz de Landazuri I, Vlagea A, García AP, et al. Primary immunodeficiency and chronic mucocutaneous candidiasis: Pathophysiological, diagnostic, and therapeutic approaches. Allergol Immunopathol (Madr). 2021; 49(1):118-27. [DOI:10.15586/aei.v49i1.20] [PMID]
- Lin Y, Slight SR, Khader SA. Th17 cytokines and vaccine-induced immunity. Semin Immunopathol. 2010; 32(1):79-90. [DOI:10.1007/s00281-009-0191-2] [PMID]
- Zimmerman O, Olbrich P, Freeman AF, Rosen LB, Uzel G, Zerbe CS, et al. STAT1 Gain-of-function mutations cause high total STAT1 levels with normal dephosphorylation. Front Immunol. 2019; 10:1433. [DOI:10.3389/fimmu.2019.01433] [PMID]
- Vandebriel RJ, Gremmer ER, van Hartskamp M, Dormans JA, Mooi FR. Effects of a diphtheria-tetanus-acellular pertussis vaccine on immune responses in murine local lymph node and lung allergy models. Clin Vaccine Immunol. 2007; 14(3):211-9. [DOI:10.1128/CVI.00306-06] [PMID]
- Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H, et al. Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol. 2008; 128(11):2640-5. [DOI:10.1038/jid.2008.139] [PMID]
- Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 2013; 39(4):676-86. [DOI:10.1016/j.immuni.2013.09.002] [PMID]
- Carey B, Lambourne J, Porter S, Hodgson T. Chronic mucocutaneous candidiasis due to gain-of-function mutation in STAT 1. Oral Dis. 2019; 25(3):684-92. [DOI:10.1111/odi.12881] [PMID]
- Scott O, Visuvanathan S, Reddy E, Mahamed D, Gu B, Roifman CM, et al. The human Stat1 gain-of-function T385M mutation causes expansion of activated T-follicular helper/T-helper 1-like CD4 T cells and sex-biased autoimmunity in specific pathogen-free mice. Front Immunol. 2023; 14:1183273. [DOI:10.3389/fimmu.2023.1183273] [PMID]
- Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017; 18(4):374-84. [DOI:10.1038/ni.3691] [PMID]
- Delsing CE, Bleeker-Rovers CP, van de Veerdonk FL, Tol J, van der Meer JW, et al. Association of esophageal candidiasis and squamous cell carcinoma. Med Mycol Case Rep. 2012; 1(1):5-8. [DOI:10.1016/j.mmcr.2012.02.003] [PMID]
- Mohd Bakri M, Mohd Hussaini H, Rachel Holmes A, David Cannon R, Mary Rich A. Revisiting the association between candidal infection and carcinoma, particularly oral squamous cell carcinoma. J Oral Microbiol. 2010; 2. [DOI:10.3402/jom.v2i0.5780] [PMID]
- Haidry RJ, Butt MA, Dunn J, Banks M, Gupta A, Smart H, et al. Radiofrequency ablation for early oesophageal squamous neoplasia: Outcomes form United Kingdom registry. World J Gastroenterol. 2013; 19(36):6011-9. [DOI:10.3748/wjg.v19.i36.6011] [PMID]
- Dotta L, Todaro F, Baronio M, Giacomelli M, Pinelli M, Giambarda M, et al. Patients with STAT1 gain-of-function mutations display increased apoptosis which is reversed by the JAK inhibitor ruxolitinib. J Clin Immunol. 2024; 44(4):85. [DOI:10.1007/s10875-024-01684-y] [PMID]
- Markham A. Baricitinib: First global approval. Drugs. 2017; 77(6):697-704. [DOI:10.1007/s40265-017-0723-3] [PMID]
- Schussler E, Beasley MB, Maglione PJ. Lung disease in primary antibody deficiencies. J Allergy Clin Immunol Pract. 2016; 4(6):1039-52. [DOI:10.1016/j.jaip.2016.08.005] [PMID]
Type of Study: Case Report and Review of Literature |
Subject:
Immunology
Received: 2025/09/28 | Accepted: 2026/02/8 | Published: 2026/04/3
Received: 2025/09/28 | Accepted: 2026/02/8 | Published: 2026/04/3
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
