
Fri, Apr 19, 2024

Volume 8, Issue 2 (4-2020)
J. Pediatr. Rev 2020, 8(2): 121-126 |
Back to browse issues page

Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Baghbanian S M, Naser Moghadasi A, Nasehi M M. Pediatric Neuromyelitis Optica Spectrum Disorders: Three Case Reports and Review of Literature. J. Pediatr. Rev 2020; 8 (2) :121-126
URL: http://jpr.mazums.ac.ir/article-1-248-en.html
URL: http://jpr.mazums.ac.ir/article-1-248-en.html



1- Department of Neurology, Bu-Ali Sina Hospital, Mazandaran University of Medical Sciences, Sari, Iran. , sm.baghbanian@mazums.ac.ir
2- MS Research Centre, Neuroscience Institute, Tehran University of Medical Sciences, Tehran, Iran.
3- Pediatric Neurology Research Center, Research Institute for Children Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2- MS Research Centre, Neuroscience Institute, Tehran University of Medical Sciences, Tehran, Iran.
3- Pediatric Neurology Research Center, Research Institute for Children Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Full-Text [PDF 436 kb]
(2070 Downloads)
| Abstract (HTML) (4375 Views)
Full-Text: (1149 Views)
1. Introduction
Neuromyelitis Optica Spectrum Disorder (NMOSD) is a rare autoimmune inflammatory and demyelinating disease of the Central Nervous System (CNS) (1). NMOSD with aquaporin-4 (AQP4)-IgG is characterized by clinical syndromes of the optic nerve, area postrema, spinal cord, brainstem, cerebral, or diencephalic presentations, or MRI findings related to these regions (1). Additional MRI findings are required to enhance the specificity of criteria for the diagnosis of NMOSD without AQP4-IgG. The Pediatric working group members noted that adult consensus criteria are applicable in pediatric patients (1). In the majority of cases, an autoantibody to the astrocytic water channel protein, AQP4, causes humoral inflammatory demyelination and leads to axonal damage (2). High relapse rate and cumulative problems due to Neuromyelitis Optica (NMO) may cause severe neurological disabilities, like permanent blindness and paralysis, which could be partly controlled by using proper attack prevention (2).
NMOSD median age onset is 39 years (3, 4). Early onset is defined as the disease presentation under the age of 17 years (5). The mean age of child NMOSD is 12-14 years and the youngest reported case was 2 years old (6). The differentiation of NMO from MS and Acute Disseminated Encephalomyelitis (ADEM) is vital for choosing an appropriate therapy. It has also been reported that some MS therapies could aggravate NMOSD and lead to permanent disability (7-9). This study aims to present three cases of pediatric NMOSD and review the other reports to emphasize that this rare disabling life-threatening disorder can be controlled with disease-modifying therapies.
2. Case Presentations
Case 1
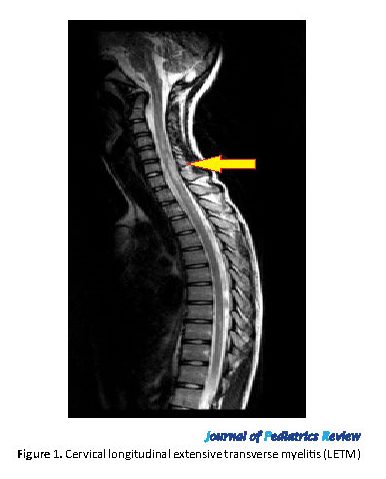
A 16-year-old female patient referred to Sina Hospital MS Clinic because of two distinct episodes of painful blurred vision with 3 months apart in 2017. Both episodes were diagnosed as optic neuritis and were recovered by pulse therapy. Neurologic examination showed bilateral mild optic atrophy, generalized hyperreflexia, and bilateral Babinski sign. AQP4-Ab and MOG (myelin-associated glycoprotein) examinations were negative. Vasculitis tests were normal. Brain MRI showed posterior corpus callosum involvement, and cervical MRI showed LETM (longitudinally extensive transverse myelitis). According to the disease criteria, she was diagnosed with NMOSD without AQP4-Ab. Azathioprine was started and her neurologic state is stable now (Figure 1).
Case 2
An 11-year-old female patient presented to Sina Hospital MS Clinic with severe right painful optic neuritis in 2016. The visual acuity reduced to 2-m finger count and right Marcus Gunn pupil. Brain MRI showed nonspecific T2 hyperintense deep white matter abnormality. AQP4-Ab was positive. She did not respond to IV steroids well, and plasma exchange was performed with acceptable recovery. She was diagnosed with NMOSD with AQP4-Ab. Azathioprine was started for her. She experienced left severe optic neuritis 3 months later. This new attack also recovered with pulse and plasmapheresis. Because of a new attack in spite of using azathioprine, rituximab was started for her. Her neurologic state is still stable.
Neuromyelitis Optica Spectrum Disorder (NMOSD) is a rare autoimmune inflammatory and demyelinating disease of the Central Nervous System (CNS) (1). NMOSD with aquaporin-4 (AQP4)-IgG is characterized by clinical syndromes of the optic nerve, area postrema, spinal cord, brainstem, cerebral, or diencephalic presentations, or MRI findings related to these regions (1). Additional MRI findings are required to enhance the specificity of criteria for the diagnosis of NMOSD without AQP4-IgG. The Pediatric working group members noted that adult consensus criteria are applicable in pediatric patients (1). In the majority of cases, an autoantibody to the astrocytic water channel protein, AQP4, causes humoral inflammatory demyelination and leads to axonal damage (2). High relapse rate and cumulative problems due to Neuromyelitis Optica (NMO) may cause severe neurological disabilities, like permanent blindness and paralysis, which could be partly controlled by using proper attack prevention (2).
NMOSD median age onset is 39 years (3, 4). Early onset is defined as the disease presentation under the age of 17 years (5). The mean age of child NMOSD is 12-14 years and the youngest reported case was 2 years old (6). The differentiation of NMO from MS and Acute Disseminated Encephalomyelitis (ADEM) is vital for choosing an appropriate therapy. It has also been reported that some MS therapies could aggravate NMOSD and lead to permanent disability (7-9). This study aims to present three cases of pediatric NMOSD and review the other reports to emphasize that this rare disabling life-threatening disorder can be controlled with disease-modifying therapies.
2. Case Presentations
Case 1
A 16-year-old female patient referred to Sina Hospital MS Clinic because of two distinct episodes of painful blurred vision with 3 months apart in 2017. Both episodes were diagnosed as optic neuritis and were recovered by pulse therapy. Neurologic examination showed bilateral mild optic atrophy, generalized hyperreflexia, and bilateral Babinski sign. AQP4-Ab and MOG (myelin-associated glycoprotein) examinations were negative. Vasculitis tests were normal. Brain MRI showed posterior corpus callosum involvement, and cervical MRI showed LETM (longitudinally extensive transverse myelitis). According to the disease criteria, she was diagnosed with NMOSD without AQP4-Ab. Azathioprine was started and her neurologic state is stable now (Figure 1).
Case 2
An 11-year-old female patient presented to Sina Hospital MS Clinic with severe right painful optic neuritis in 2016. The visual acuity reduced to 2-m finger count and right Marcus Gunn pupil. Brain MRI showed nonspecific T2 hyperintense deep white matter abnormality. AQP4-Ab was positive. She did not respond to IV steroids well, and plasma exchange was performed with acceptable recovery. She was diagnosed with NMOSD with AQP4-Ab. Azathioprine was started for her. She experienced left severe optic neuritis 3 months later. This new attack also recovered with pulse and plasmapheresis. Because of a new attack in spite of using azathioprine, rituximab was started for her. Her neurologic state is still stable.
Case 3
A 13-year-old female patient was consulted with a history of cervical pain, and urinary retention ended in quadriparesis, which was improved by pulse therapy in 2017. She also had another relapse with ataxia and upper limb pain, which was recovered by pulse therapy. Neurological examination showed signs of upper motor neuron lesions. Vasculitis test was normal, and high titer AQP4-Ab was reported. Azathioprine was administered, and she experienced no relapse since then. Cervical MRI showed LETM, but brain MRI did not show any abnormality.
3. Discussion and review of the literature
According to recent consensus diagnostic criteria, NMOSD is categorized into NMOSD with AQP4-Ab and NMOSD without AQP4-Ab (10). In these criteria, 6 core clinical characteristics have been defined each implicates one area in the central nervous system: optic nerve, spinal cord, postrema area of the dorsal medulla, diencephalon, brainstem, and cerebral hemispheres. One core criteria with positive AQP4-Ab is defined as NMOSD with AQP4-Ab. However, in NMOSD without AQP4-Ab, the patients must present 2 or more different core clinical characteristics, and at least one core criterion must include optic neuritis, transverse myelitis, or postrema clinical syndrome (10). Consensus criteria recommend NMO-Ab detection with cell base assay, due to its high sensitivity (76.7%) and low false positivity (0.1%) in adult patients. However, the cell-based assay is not available in most medical clinics so far. Also, the sensitivity of indirect immunofluorescence and ELISA assay is not low (mean sensitivity is 63%–64% for each) in adult patients (11).
Longitudinally Extended Transverse Myelitis (LETM) in the spinal cord MRI of a patient with transverse myelitis is the most specific and highly suggested neuroimaging finding in NMOSD. This lesion involves cord central gray matter and is associated with cord expansion of more than 2/3 of the axial cord section, extending more than 3 vertebral bodies, which is enhanced with gadolinium. Nevertheless, acute myelitis in NMOSD is not always associated with LETM, and 7%-14% of NMOSD cases initially present with short myelitis (12). Normal brain MRI is a key supportive finding in NMOSD. About 60% of NMOSD patients show nonspecific asymptomatic white matter lesions, and up to 16% of them show complete Barkhof MS criteria (13). Gender does not seem to predict the frequency of attacks once the relapsing disease is presented (5). On the other hand, ADEM like presentation is common in pediatric NMOSD (14, 15).
Most clinical, laboratory, and neuroimaging characteristics of pediatric NMOSD are comparable to those of adult NMOSD, and according to the pediatric NMOSD working group, the same criteria could be used for pediatric NMOSD. New diagnostic criteria for neuromyelitis optica spectrum disease markedly resolved diagnostic challenges, especially in seronegative NMO-Ab circumstances (16). The pediatric onset of NMOSD accounts for 4% of NMOSD seropositive cases. The female/male sex ratio is reported as 9:1 in NMOSD compared to 3:1 in MS. In North America, African American children dominate 33%-34% of pediatric NMOSD cases, then Caucasians, Latin American, white non-Caucasian, and finally, Asians are placed (17).
According to the US Network of Pediatric MS Centers, 38 cases of pediatric NMOSD show some demographic and seropositive characteristics. In this report, the Mean±SD age of onset is 10.2±4.7 years in NMOSD, and 65% of NMOSD cohort is reported seropositive for AQP4-Ab, which is close to reports about adults, but 32% of patients were male in this case series, and F/M ratio was 1.5:1 for patients with NMOSD who were under 11 years old (18). About 19.3 years studying on 12 pediatric NMO patients showed no difference between the adult NMO and pediatric NMO patients in the number of brain MRI lesions (5).
In a study on 58 cases of pediatric NMOSD, the median age at symptom onset was reported 12 years (range: 4-18 years), and 98% presented attacks of either optic neuritis (83%) or transverse myelitis (78%) or both, 45% had frequent cerebral symptoms (encephalopathy, ataxia, ophthalmoparesis, intractable vomiting, seizures, or hiccups), 68% had brain MRI abnormalities, 76% showed other autoantibodies, and 42% had a coexisting autoimmune disorder. The frequency of AQP4-Ab in children with inflammatory disorders of the CNS was 78% for relapsing NMOSD, 20% for partial forms of NMOSD, and 66% fulfilled 2006 NMOSD diagnostic criteria. Azathioprine was administered as monotherapy for 6 patients, and 8 patients received rituximab, and only one patient was reported as relapse-free after starting rituximab (19).
In a study evaluating 118 pediatric patients, only 6 patients fulfilled diagnostic criteria of NMOSD, and just one of them was seropositive for AQP4-Ab. The authors concluded that NMOSD is rare in the white European pediatric population (20). Follow up of 29 pediatric NMOSD patients series with the average age of 13 years, 3/1 female/male ratio, and 92% seropositive for anti-aquaporin-4 at the disease onset showed that early onset of NMO morbidity is higher than MS so that NMOSD may have a negative influence on schooling. The authors emphasized that the most important interference factor in the pediatric NMOSD diagnosis is pediatric MS, and Intraneural Facilitation (INF) therapy may aggravate disease status in NMOSD patients (21).
In a follow-up of 17 pediatric NMOSD patients, only 8 of them (47%) were AQP4-Ab seropositive, and 78% had relapsing NMOSD. It means that 31% of relapsing NMOSD were seronegative; therefore, we could explain why NMOSD could present as relapsing disease without AQP4-Ab. In this study, relapsing NMOSD in seropositive patients is more common than monophasic NMOSD. However, there was no difference between seropositive and seronegative patients with respect to their sex and ethnicity. About 53% of these patients, similar to adult NMOSD patients, had brain MRI abnormality, and the most common lesions were brainstem lesions extending from medulla to the spinal cord (22).
In the evaluation of 6 pediatric NMOSD patients, their median age was 11 years at the time of the study, and the F/M ratio was 5/1. Also, all cases presented bilateral optic neuritis, 66% had abnormal brain MRI from the onset, and 50% had symptoms associated with the brain lesions during their disease. AQP4-Ab seropositivity was reported in 80% of the patients. Optic neuropathy was the most impaired feature. TM was presented in all the patients during the disorder (23). This Latin American case series emphasized that the race might be a noticeable demographic variable for NMOSD in children (23).
In a cohort study belongs to UK national pediatric onset of NMOSD, 20 pediatric NMO with the median age of 10.5 years (range: 2.9-16.8 years) met the inclusion criteria. About 90% of them were female, 60% were AQP4-Ab positive, 12% CSF OCB positive, 90% experienced relapsing course, 75% with MRI brain abnormalities, 20% had Barkhof space involvement, and 100% reported LETM. The most common brain MRI lesion (53%) location was centrum semiovale/deep white matter. Unilateral optic neuritis was the first common presentation (40%). This study has emphasized that the pediatric NMO first presentation might be similar to other acquired demyelinating syndromes, and careful attention to distinct MRI brain lesion features is very helpful to minimize the risk of incorrect diagnosis, and also prevent early recurrence and visual disability of pediatric NMO (24).
In a study on the Korean children, 3 out of 21 patients met the international NMO criteria. The first case was a 7 years old female who showed bilateral optic neuritis. Brain MRI showed the involvement of corpus callosum, thalamus, and periventricular area. Spinal MRI revealed LETM of the thoracic region. She died at the age of 14 years because of pneumonia. The second patient was a 7 years old male child presented with isolated transverse myelitis, which bilateral optic neuritis added to his clinical presentation 3 months later. His brain MRI showed subcortical T2-weighted hyperintensity in temporal and occipital lobes. And the last case was an 11.7 years old female patient presented with transverse myelitis accompanied by sensory signs and the left eye visual impairment followed by four clinical relapses of transverse myelitis and optic neuritis (25).
In a Brazilian study, 11 pediatric NMOSD patients were under study. F/M ratio was 2.6/1, AQP4-IgG was positive in 72.7% of patients, and the mean age of disease onset was reported as 14 years. MRI showed cervical LETM in all patients, and also brain lesions in 45.5% of patients. All of the patients tolerated azathioprine (26).
The current approach in treatment with rituximab is a step-up approach, which reserves rituximab for refractory cases (27). Accordingly, some studies suggest rituximab as a first line of maintenance therapy in pediatric NMOSD (28, 29). In a longitudinal follow-up of 114 adult NMOSD patients, 10 pediatric NMOSD patients were defined retrospectively, whose treatment escalated to rituximab from azathioprine because of severe and recurrent new relapses in half of them (29).
Current practice in rituximab administration is a repeated fixed time infusion every 6-12 months but recently proposed approach suggests personalized treatment according to regular B-cell depletion and reconstitution monitoring (27, 30). In conclusion, pediatric NMOSD is a rare but life-threatening disease, which pediatricians and pediatric neurologists must be aware of its presentations and treatment.
Ethical Considerations
Compliance with ethical guidelines
The study was conducted upon agreement of the Ethics Committee of Tehran University of Medical Sciences, and the inform consent of the patients is available upon request.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors contributions
All authors contributed in preparing this article.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgements
The authors would like to thank Sina Hospital Neuromyelitis Optica Spectrum Disease Cohort Study Group of Tehran University of Medical Science for providing medical records and questionnaire files.
References
Wingerchuk DM, Weinshenker BG. Neuromyelitis optica (Devic’s syndrome). Handbook of Clinical Neurology. 2014; 122:581-99. [DOI:10.1016/B978-0-444-52001-2.00025-X] [PMID]
Wegner C. Recent insights into the pathology of multiple sclerosis and neuromyelitis optica. Clinical Neurology and Neurosurgery. 2013; 115(Suppl. 1):S38-41. [DOI:10.1016/j.clineuro.2013.09.019] [PMID]
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999; 53(5):1107-14. [DOI:10.1212/WNL.53.5.1107] [PMID]
Kantarci OH, Weinshenker BG. Natural history of multiple sclerosis. Neurologic Clinics. 2005; 23(1):17-38. [DOI:10.1016/j.ncl.2004.10.002] [PMID]
McKeon A, Lennon VA, Lotze T, Tenenbaum S, Ness JM, Rensel M, et al. CNS aquaporin-4 autoimmunity in children. Neurology. 2008; 71(2):93-100. [DOI:10.1212/01.wnl.0000314832.24682.c6] [PMID]
Yuksel D, Senbil N, Yilmaz D, Yavuz Gurer YK. Devic’s neuromyelitis optica in an infant case. Journal of Child Neurology. 2007; 22(9):1143-6. [DOI:10.1177/0883073807306270] [PMID]
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Multiple Sclerosis Journal. 2012; 18(10):1480-3. [DOI:10.1177/1352458512439439] [PMID]
Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Archives of Neurology. 2012; 69(2):239-45. [DOI:10.1001/archneurol.2011.216] [PMID]
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Multiple Sclerosis Journal. 2012; 18(1):113-5. [DOI:10.1177/1352458511431973] [PMID]
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015; 85(2):177-89. [DOI:10.1212/WNL.0000000000001729] [PMID] [PMCID]
Jarius S, Wildemann B. Aquaporin-4 antibodies (NMOIgG) as a serological marker of neuromyelitis optica: A critical review of the literature. Brain Pathology. 2013; 23(6):661-83. [DOI:10.1111/bpa.12084] [PMID]
Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurology. 2015; 72(1):81-7. [DOI:10.1001/jamaneurol.2014.2137] [PMID] [PMCID]
Kim W, Park MS, Lee SH, Kim SH, Jung IJ, Takahashi T, et al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Multiple Sclerosis Journal. 2010; 16(10):1229-36. [DOI:10.1177/1352458510376640] [PMID]
Zare-Shahabadi A, Langroodi HG, Azimi AR, Sahraian MA, Harirchian MH, Baghbanian SM. Neuromyelitis optica and pregnancy. Acta Neurologica Belgica. 2016; 116(4):431-8. [DOI:10.1007/s13760-016-0654-x] [PMID]
Krupp LB, Tardieu M, Amato MP, Banwell B, Chitnis T, Dale RC, et al. International pediatric multiple sclerosis study group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: Revisions to the 2007 definitions. Multiple Sclerosis Journal. 2013; 19(10):1261-7. [DOI:10.1177/1352458513484547] [PMID]
Hyun JW, Jeong IH, Joung A, Kim SH, Kim HJ. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016; 86(19):1772-9. [DOI:10.1212/WNL.0000000000002655] [PMID]
Lotze TE, Northrop JL, Hutton GJ, Ross B, Schiffman JS, Hunter JV. Spectrum of pediatric neuromyelitis optica. Pediatrics. 2008; 122(5):e1039-47 [DOI:10.1542/peds.2007-2758] [PMID]
Chitnis T, Ness J, Krupp L, Waubant E, Hunt T, Olsen CS, et al. Clinical features of neuromyelitis optica in children: US network of pediatric ms centers report. Neurology. 2016; 86(3):245-52. [DOI:10.1212/WNL.0000000000002283] [PMID] [PMCID]
Collongues N, Marignier R, Zéphir H, Papeix C, Fontaine B, Blanc F, et al. Long-term follow-up of neuromyelitis optica with a pediatric onset. Neurology. 2010; 75(12):1084-8 [DOI:10.1212/WNL.0b013e3181f39a66] [PMID]
Huppke P, Blüthner M, Bauer O, Stark W, Reinhardt K, Huppke B, et al. Neuromyelitis optica and NMO-IgG in European pediatric patients. Neurology. 2010; 75(19):1740-4. [DOI:10.1212/WNL.0b013e3181fc2823] [PMID]
Fragoso YD, Ferreira ML, Oliveira EM, Domingues RB, Ribeiro TA, Brooks JB, et al. Neuromyelitis optica with onset in childhood and adolescence. Pediatric Neurology. 2014; 50(1):66-8. [DOI:10.1016/j.pediatrneurol.2013.07.003] [PMID]
Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar-Or A, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008; 70(5):344-52. [DOI:10.1212/01.wnl.0000284600.80782.d5] [PMID]
Peña JA1, Ravelo ME, Mora-La Cruz E, Montiel-Nava C. NMO in pediatric patients: Brain involvement and clinical expression. Archives of Neuro-Psychiatry. 2011; 69(1):34-8. [DOI:10.1590/S0004-282X2011000100008] [PMID]
Absoud M, Lim MJ, Appleton R, Jacob A, Kitley J, Leite MI, et al. Paediatric neuromyelitis optica: Clinical, MRI of the brain and prognostic features. Journal of Neurology, Neurosurgery, and Psychiatry. 2015; 86(4):470-2. [DOI:10.1136/jnnp-2014-308550] [PMID]
Lim BC, Hwang H, Kim KJ, Hwang YS, Cheon JE, Kim IO, et al. Relapsing demyelinating CNS disease in a Korean pediatric population: Multiple sclerosis versus neuromyelitis optica. Multiple Sclerosis Journal. 2011; 17(1):67-73. [DOI:10.1177/1352458510382685] [PMID]
Fragomeni MO, Bichuetti DB, Oliveira EML. Pediatric-onset multiple sclerosis in Brazilian patients: Clinical features, treatment response and comparison to pediatric neuromyelitis optica spectrum disorders. Multiple Sclerosis and Related Disorders. 2018; 25:138-42 [DOI:10.1016/j.msard.2018.07.036] [PMID]
Baghbanian SM, Asgari N, Sahraian MA, Moghadasi AN. A comparison of pediatric and adult neuromyelitis optica spectrum disorders: A review of clinical manifestation, diagnosis, and treatment. Journal of the Neurological Sciences. 2018; 388:222-31. [DOI:10.1016/j.jns.2018.02.028] [PMID]
Olivieri G, Nociti V, Iorio R, Stefanini MC, Losavio FA, Mirabella M, et al. Rituximab as a first-line treatment in pediatric neuromyelitis optica spectrum disorder. Neurological Sciences. 2015; 36(12):2301-2. [DOI:10.1007/s10072-015-2368-x] [PMID]
Baghbanian SM, Sahraian MA, Moghadasi AN, Asgari N. Disability and therapeutic response in paediatric neuromyelitis optica spectrum disorder: A case series from Iran. Iranian Journal of Child Neurology. 2019; 13(3):99-104. [DOI:10.22037/ijcn.v13i3.22715]
Nosadini M, Alper G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S, et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurology Neuroimmunology & Neuroinflammation. 2016; 3(1):e188. [DOI:10.1212/NXI.0000000000000188] [PMID] [PMCID]
A 13-year-old female patient was consulted with a history of cervical pain, and urinary retention ended in quadriparesis, which was improved by pulse therapy in 2017. She also had another relapse with ataxia and upper limb pain, which was recovered by pulse therapy. Neurological examination showed signs of upper motor neuron lesions. Vasculitis test was normal, and high titer AQP4-Ab was reported. Azathioprine was administered, and she experienced no relapse since then. Cervical MRI showed LETM, but brain MRI did not show any abnormality.
3. Discussion and review of the literature
According to recent consensus diagnostic criteria, NMOSD is categorized into NMOSD with AQP4-Ab and NMOSD without AQP4-Ab (10). In these criteria, 6 core clinical characteristics have been defined each implicates one area in the central nervous system: optic nerve, spinal cord, postrema area of the dorsal medulla, diencephalon, brainstem, and cerebral hemispheres. One core criteria with positive AQP4-Ab is defined as NMOSD with AQP4-Ab. However, in NMOSD without AQP4-Ab, the patients must present 2 or more different core clinical characteristics, and at least one core criterion must include optic neuritis, transverse myelitis, or postrema clinical syndrome (10). Consensus criteria recommend NMO-Ab detection with cell base assay, due to its high sensitivity (76.7%) and low false positivity (0.1%) in adult patients. However, the cell-based assay is not available in most medical clinics so far. Also, the sensitivity of indirect immunofluorescence and ELISA assay is not low (mean sensitivity is 63%–64% for each) in adult patients (11).
Longitudinally Extended Transverse Myelitis (LETM) in the spinal cord MRI of a patient with transverse myelitis is the most specific and highly suggested neuroimaging finding in NMOSD. This lesion involves cord central gray matter and is associated with cord expansion of more than 2/3 of the axial cord section, extending more than 3 vertebral bodies, which is enhanced with gadolinium. Nevertheless, acute myelitis in NMOSD is not always associated with LETM, and 7%-14% of NMOSD cases initially present with short myelitis (12). Normal brain MRI is a key supportive finding in NMOSD. About 60% of NMOSD patients show nonspecific asymptomatic white matter lesions, and up to 16% of them show complete Barkhof MS criteria (13). Gender does not seem to predict the frequency of attacks once the relapsing disease is presented (5). On the other hand, ADEM like presentation is common in pediatric NMOSD (14, 15).
Most clinical, laboratory, and neuroimaging characteristics of pediatric NMOSD are comparable to those of adult NMOSD, and according to the pediatric NMOSD working group, the same criteria could be used for pediatric NMOSD. New diagnostic criteria for neuromyelitis optica spectrum disease markedly resolved diagnostic challenges, especially in seronegative NMO-Ab circumstances (16). The pediatric onset of NMOSD accounts for 4% of NMOSD seropositive cases. The female/male sex ratio is reported as 9:1 in NMOSD compared to 3:1 in MS. In North America, African American children dominate 33%-34% of pediatric NMOSD cases, then Caucasians, Latin American, white non-Caucasian, and finally, Asians are placed (17).
According to the US Network of Pediatric MS Centers, 38 cases of pediatric NMOSD show some demographic and seropositive characteristics. In this report, the Mean±SD age of onset is 10.2±4.7 years in NMOSD, and 65% of NMOSD cohort is reported seropositive for AQP4-Ab, which is close to reports about adults, but 32% of patients were male in this case series, and F/M ratio was 1.5:1 for patients with NMOSD who were under 11 years old (18). About 19.3 years studying on 12 pediatric NMO patients showed no difference between the adult NMO and pediatric NMO patients in the number of brain MRI lesions (5).
In a study on 58 cases of pediatric NMOSD, the median age at symptom onset was reported 12 years (range: 4-18 years), and 98% presented attacks of either optic neuritis (83%) or transverse myelitis (78%) or both, 45% had frequent cerebral symptoms (encephalopathy, ataxia, ophthalmoparesis, intractable vomiting, seizures, or hiccups), 68% had brain MRI abnormalities, 76% showed other autoantibodies, and 42% had a coexisting autoimmune disorder. The frequency of AQP4-Ab in children with inflammatory disorders of the CNS was 78% for relapsing NMOSD, 20% for partial forms of NMOSD, and 66% fulfilled 2006 NMOSD diagnostic criteria. Azathioprine was administered as monotherapy for 6 patients, and 8 patients received rituximab, and only one patient was reported as relapse-free after starting rituximab (19).
In a study evaluating 118 pediatric patients, only 6 patients fulfilled diagnostic criteria of NMOSD, and just one of them was seropositive for AQP4-Ab. The authors concluded that NMOSD is rare in the white European pediatric population (20). Follow up of 29 pediatric NMOSD patients series with the average age of 13 years, 3/1 female/male ratio, and 92% seropositive for anti-aquaporin-4 at the disease onset showed that early onset of NMO morbidity is higher than MS so that NMOSD may have a negative influence on schooling. The authors emphasized that the most important interference factor in the pediatric NMOSD diagnosis is pediatric MS, and Intraneural Facilitation (INF) therapy may aggravate disease status in NMOSD patients (21).
In a follow-up of 17 pediatric NMOSD patients, only 8 of them (47%) were AQP4-Ab seropositive, and 78% had relapsing NMOSD. It means that 31% of relapsing NMOSD were seronegative; therefore, we could explain why NMOSD could present as relapsing disease without AQP4-Ab. In this study, relapsing NMOSD in seropositive patients is more common than monophasic NMOSD. However, there was no difference between seropositive and seronegative patients with respect to their sex and ethnicity. About 53% of these patients, similar to adult NMOSD patients, had brain MRI abnormality, and the most common lesions were brainstem lesions extending from medulla to the spinal cord (22).
In the evaluation of 6 pediatric NMOSD patients, their median age was 11 years at the time of the study, and the F/M ratio was 5/1. Also, all cases presented bilateral optic neuritis, 66% had abnormal brain MRI from the onset, and 50% had symptoms associated with the brain lesions during their disease. AQP4-Ab seropositivity was reported in 80% of the patients. Optic neuropathy was the most impaired feature. TM was presented in all the patients during the disorder (23). This Latin American case series emphasized that the race might be a noticeable demographic variable for NMOSD in children (23).
In a cohort study belongs to UK national pediatric onset of NMOSD, 20 pediatric NMO with the median age of 10.5 years (range: 2.9-16.8 years) met the inclusion criteria. About 90% of them were female, 60% were AQP4-Ab positive, 12% CSF OCB positive, 90% experienced relapsing course, 75% with MRI brain abnormalities, 20% had Barkhof space involvement, and 100% reported LETM. The most common brain MRI lesion (53%) location was centrum semiovale/deep white matter. Unilateral optic neuritis was the first common presentation (40%). This study has emphasized that the pediatric NMO first presentation might be similar to other acquired demyelinating syndromes, and careful attention to distinct MRI brain lesion features is very helpful to minimize the risk of incorrect diagnosis, and also prevent early recurrence and visual disability of pediatric NMO (24).
In a study on the Korean children, 3 out of 21 patients met the international NMO criteria. The first case was a 7 years old female who showed bilateral optic neuritis. Brain MRI showed the involvement of corpus callosum, thalamus, and periventricular area. Spinal MRI revealed LETM of the thoracic region. She died at the age of 14 years because of pneumonia. The second patient was a 7 years old male child presented with isolated transverse myelitis, which bilateral optic neuritis added to his clinical presentation 3 months later. His brain MRI showed subcortical T2-weighted hyperintensity in temporal and occipital lobes. And the last case was an 11.7 years old female patient presented with transverse myelitis accompanied by sensory signs and the left eye visual impairment followed by four clinical relapses of transverse myelitis and optic neuritis (25).
In a Brazilian study, 11 pediatric NMOSD patients were under study. F/M ratio was 2.6/1, AQP4-IgG was positive in 72.7% of patients, and the mean age of disease onset was reported as 14 years. MRI showed cervical LETM in all patients, and also brain lesions in 45.5% of patients. All of the patients tolerated azathioprine (26).
The current approach in treatment with rituximab is a step-up approach, which reserves rituximab for refractory cases (27). Accordingly, some studies suggest rituximab as a first line of maintenance therapy in pediatric NMOSD (28, 29). In a longitudinal follow-up of 114 adult NMOSD patients, 10 pediatric NMOSD patients were defined retrospectively, whose treatment escalated to rituximab from azathioprine because of severe and recurrent new relapses in half of them (29).
Current practice in rituximab administration is a repeated fixed time infusion every 6-12 months but recently proposed approach suggests personalized treatment according to regular B-cell depletion and reconstitution monitoring (27, 30). In conclusion, pediatric NMOSD is a rare but life-threatening disease, which pediatricians and pediatric neurologists must be aware of its presentations and treatment.
Ethical Considerations
Compliance with ethical guidelines
The study was conducted upon agreement of the Ethics Committee of Tehran University of Medical Sciences, and the inform consent of the patients is available upon request.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors contributions
All authors contributed in preparing this article.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgements
The authors would like to thank Sina Hospital Neuromyelitis Optica Spectrum Disease Cohort Study Group of Tehran University of Medical Science for providing medical records and questionnaire files.
References
Wingerchuk DM, Weinshenker BG. Neuromyelitis optica (Devic’s syndrome). Handbook of Clinical Neurology. 2014; 122:581-99. [DOI:10.1016/B978-0-444-52001-2.00025-X] [PMID]
Wegner C. Recent insights into the pathology of multiple sclerosis and neuromyelitis optica. Clinical Neurology and Neurosurgery. 2013; 115(Suppl. 1):S38-41. [DOI:10.1016/j.clineuro.2013.09.019] [PMID]
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999; 53(5):1107-14. [DOI:10.1212/WNL.53.5.1107] [PMID]
Kantarci OH, Weinshenker BG. Natural history of multiple sclerosis. Neurologic Clinics. 2005; 23(1):17-38. [DOI:10.1016/j.ncl.2004.10.002] [PMID]
McKeon A, Lennon VA, Lotze T, Tenenbaum S, Ness JM, Rensel M, et al. CNS aquaporin-4 autoimmunity in children. Neurology. 2008; 71(2):93-100. [DOI:10.1212/01.wnl.0000314832.24682.c6] [PMID]
Yuksel D, Senbil N, Yilmaz D, Yavuz Gurer YK. Devic’s neuromyelitis optica in an infant case. Journal of Child Neurology. 2007; 22(9):1143-6. [DOI:10.1177/0883073807306270] [PMID]
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Multiple Sclerosis Journal. 2012; 18(10):1480-3. [DOI:10.1177/1352458512439439] [PMID]
Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Archives of Neurology. 2012; 69(2):239-45. [DOI:10.1001/archneurol.2011.216] [PMID]
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Multiple Sclerosis Journal. 2012; 18(1):113-5. [DOI:10.1177/1352458511431973] [PMID]
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015; 85(2):177-89. [DOI:10.1212/WNL.0000000000001729] [PMID] [PMCID]
Jarius S, Wildemann B. Aquaporin-4 antibodies (NMOIgG) as a serological marker of neuromyelitis optica: A critical review of the literature. Brain Pathology. 2013; 23(6):661-83. [DOI:10.1111/bpa.12084] [PMID]
Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurology. 2015; 72(1):81-7. [DOI:10.1001/jamaneurol.2014.2137] [PMID] [PMCID]
Kim W, Park MS, Lee SH, Kim SH, Jung IJ, Takahashi T, et al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Multiple Sclerosis Journal. 2010; 16(10):1229-36. [DOI:10.1177/1352458510376640] [PMID]
Zare-Shahabadi A, Langroodi HG, Azimi AR, Sahraian MA, Harirchian MH, Baghbanian SM. Neuromyelitis optica and pregnancy. Acta Neurologica Belgica. 2016; 116(4):431-8. [DOI:10.1007/s13760-016-0654-x] [PMID]
Krupp LB, Tardieu M, Amato MP, Banwell B, Chitnis T, Dale RC, et al. International pediatric multiple sclerosis study group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: Revisions to the 2007 definitions. Multiple Sclerosis Journal. 2013; 19(10):1261-7. [DOI:10.1177/1352458513484547] [PMID]
Hyun JW, Jeong IH, Joung A, Kim SH, Kim HJ. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016; 86(19):1772-9. [DOI:10.1212/WNL.0000000000002655] [PMID]
Lotze TE, Northrop JL, Hutton GJ, Ross B, Schiffman JS, Hunter JV. Spectrum of pediatric neuromyelitis optica. Pediatrics. 2008; 122(5):e1039-47 [DOI:10.1542/peds.2007-2758] [PMID]
Chitnis T, Ness J, Krupp L, Waubant E, Hunt T, Olsen CS, et al. Clinical features of neuromyelitis optica in children: US network of pediatric ms centers report. Neurology. 2016; 86(3):245-52. [DOI:10.1212/WNL.0000000000002283] [PMID] [PMCID]
Collongues N, Marignier R, Zéphir H, Papeix C, Fontaine B, Blanc F, et al. Long-term follow-up of neuromyelitis optica with a pediatric onset. Neurology. 2010; 75(12):1084-8 [DOI:10.1212/WNL.0b013e3181f39a66] [PMID]
Huppke P, Blüthner M, Bauer O, Stark W, Reinhardt K, Huppke B, et al. Neuromyelitis optica and NMO-IgG in European pediatric patients. Neurology. 2010; 75(19):1740-4. [DOI:10.1212/WNL.0b013e3181fc2823] [PMID]
Fragoso YD, Ferreira ML, Oliveira EM, Domingues RB, Ribeiro TA, Brooks JB, et al. Neuromyelitis optica with onset in childhood and adolescence. Pediatric Neurology. 2014; 50(1):66-8. [DOI:10.1016/j.pediatrneurol.2013.07.003] [PMID]
Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar-Or A, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008; 70(5):344-52. [DOI:10.1212/01.wnl.0000284600.80782.d5] [PMID]
Peña JA1, Ravelo ME, Mora-La Cruz E, Montiel-Nava C. NMO in pediatric patients: Brain involvement and clinical expression. Archives of Neuro-Psychiatry. 2011; 69(1):34-8. [DOI:10.1590/S0004-282X2011000100008] [PMID]
Absoud M, Lim MJ, Appleton R, Jacob A, Kitley J, Leite MI, et al. Paediatric neuromyelitis optica: Clinical, MRI of the brain and prognostic features. Journal of Neurology, Neurosurgery, and Psychiatry. 2015; 86(4):470-2. [DOI:10.1136/jnnp-2014-308550] [PMID]
Lim BC, Hwang H, Kim KJ, Hwang YS, Cheon JE, Kim IO, et al. Relapsing demyelinating CNS disease in a Korean pediatric population: Multiple sclerosis versus neuromyelitis optica. Multiple Sclerosis Journal. 2011; 17(1):67-73. [DOI:10.1177/1352458510382685] [PMID]
Fragomeni MO, Bichuetti DB, Oliveira EML. Pediatric-onset multiple sclerosis in Brazilian patients: Clinical features, treatment response and comparison to pediatric neuromyelitis optica spectrum disorders. Multiple Sclerosis and Related Disorders. 2018; 25:138-42 [DOI:10.1016/j.msard.2018.07.036] [PMID]
Baghbanian SM, Asgari N, Sahraian MA, Moghadasi AN. A comparison of pediatric and adult neuromyelitis optica spectrum disorders: A review of clinical manifestation, diagnosis, and treatment. Journal of the Neurological Sciences. 2018; 388:222-31. [DOI:10.1016/j.jns.2018.02.028] [PMID]
Olivieri G, Nociti V, Iorio R, Stefanini MC, Losavio FA, Mirabella M, et al. Rituximab as a first-line treatment in pediatric neuromyelitis optica spectrum disorder. Neurological Sciences. 2015; 36(12):2301-2. [DOI:10.1007/s10072-015-2368-x] [PMID]
Baghbanian SM, Sahraian MA, Moghadasi AN, Asgari N. Disability and therapeutic response in paediatric neuromyelitis optica spectrum disorder: A case series from Iran. Iranian Journal of Child Neurology. 2019; 13(3):99-104. [DOI:10.22037/ijcn.v13i3.22715]
Nosadini M, Alper G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S, et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurology Neuroimmunology & Neuroinflammation. 2016; 3(1):e188. [DOI:10.1212/NXI.0000000000000188] [PMID] [PMCID]
Type of Study: Case Report and Review of Literature |
Subject:
Neurology
Received: 2019/06/22 | Accepted: 2019/08/28 | Published: 2020/04/1
Received: 2019/06/22 | Accepted: 2019/08/28 | Published: 2020/04/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Articles Copyright © The Author(s).
Owned by Mazandaran University of Medical Sciences.
Published by Negah Institute for Scientific Communication.
Journal of Pediatrics Review (JPR)
Bou Ali Sina Hospital, Pasdaran Boulevard, Sari, Iran.
Journal Tel: +98 - 1133342331
Publisher Tel: +9821 8603 6497-
+9821 8603 7228 (EXT: 104)
Website: http://jpr.mazums.ac.ir/
E-mail: jpr.mazums@gmail.com,
jpr@mazums.ac.ir
