
Sun, Jun 14, 2026


Volume 8, Issue 2 (4-2020)
J. Pediatr. Rev 2020, 8(2): 133-138 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Karami H, Ghaffari J, Moradi S, Vahedi L, Mohammadi-Kharkeshi F. Humoral and Complement Systems in Children With Acute Immune Thrombocytopenic Purpura. J. Pediatr. Rev 2020; 8 (2) :133-138
URL: http://jpr.mazums.ac.ir/article-1-236-en.html
URL: http://jpr.mazums.ac.ir/article-1-236-en.html
Hossein Karami1 

 , Javad Ghaffari *2 , Siavash Moradi3 , Laleh Vahedi4 , Fatemeh Mohammadi-Kharkeshi5
, Javad Ghaffari *2 , Siavash Moradi3 , Laleh Vahedi4 , Fatemeh Mohammadi-Kharkeshi5
, Javad Ghaffari *2 , Siavash Moradi3 , Laleh Vahedi4 , Fatemeh Mohammadi-Kharkeshi5
1- Pediatric Infectious Diseases Research Center, Mazandaran University of Medical Sciences, Sari, Iran.
2- Pediatric Infectious Diseases Research Center, Mazandaran University of Medical Sciences, Sari, Iran. ,javadneg@yahoo.com
3- Community Medicine Specialist, Education Development Center, Mazandaran University of Medical Sciences, Sari, Iran.
4- Gastrointestinal Cancer Research Center, Mazandaran University of Medical Sciences, Sari, Iran.
5- Pediatrician, Mazandaran University of Medical Sciences, Sari, Iran.
2- Pediatric Infectious Diseases Research Center, Mazandaran University of Medical Sciences, Sari, Iran. ,
3- Community Medicine Specialist, Education Development Center, Mazandaran University of Medical Sciences, Sari, Iran.
4- Gastrointestinal Cancer Research Center, Mazandaran University of Medical Sciences, Sari, Iran.
5- Pediatrician, Mazandaran University of Medical Sciences, Sari, Iran.
Full-Text [PDF 698 kb]
(2567 Downloads)
| Abstract (HTML) (7309 Views)

Full-Text: (2136 Views)
1. Introduction
Immune Thrombocytopenia (ITP) is an immune and or autoimmune disease with bleeding tendency due to accelerated platelet destruction as well as decreased platelet production mediated in part by autoantibodies. ITP has several etiologies, natural histories, and responses to treatment. In children with ITP, there is sometimes a history of recent infections. In the endemic areas, in adults with ITP, Helicobacter pylori, human immune deficiency virus, or hepatitis C are implicated (1). Vaccination can also induce ITP, such as the measles-mumps-rubella vaccine (MMR, ~1:40000). Some cases of ITP due to vaccination (hepatitis B, diphtheria-tetanus-pertussis, and hepatitis A vaccines) have been reported in children (2).
In patients with ITP, the platelet-autoreactive T cells induce interleukin-2 (IL-2) and interferon-gamma (IFN-γ) predominantly, then with alteration in regulatory T cells (Treg) number and function, a shift toward stimulatory monocytes with enhanced Fcγ receptors (FcγR)-mediated phagocytic capacity causes immune dysregulation in ITP. Some researchers reported increased Th17 cells or IL-17 cytokine in ITP patients whereas other reports did not detect any differences. Resolution in Treg function in ITP patients induces increased platelet counts after the use of these agents (3). Similar results have been reported with rituximab that improve Treg function. High-dose dexamethasone treatment increases the number of circulating Treg cells and decreases Th1 cells (4, 5).
Immunodeficiency disorders are diseases of defective innate adaptive immunity with diverse clinical phenotypes and genotypes manifested by recurrent severe infections, autoimmunity, and lymphoproliferation (6). Antibody deficiencies are more common in primary immune deficiency followed by well-defined syndromes with immunodeficiency, phagocytic disorders, complement deficiencies, immunodeficiencies affecting cellular and humoral immunity in nine patients, disease of immune dysregulation and defects in intrinsic and innate immunity (7).
In primary immune deficiency, especially in antibody disorders, chronic and recurrent infections, chronic inflammation, and autoimmunity are more common. Besides, they tend to develop malignancies and lymphoproliferative disorders. Prevalence of autoimmune disorders in Common Variable Immunodeficiency (CVID), hyper IgM (HIgM), secretory IgA deficiency (SIgAD) and X-linked agammaglobulinemia (XLA) patients were 38 (30.4%), 13 (40.6%), 14 (22.2%), and 11 (18.3%), respectively. Prevalence of ITP in CVID, HIgM, SIgAD, and XLA patients were 5 (4%), 5 (15.5%), 0 (0%), and 3 (5%), respectively (8, 9). In the present study, we explored the role of humoral and complement systems in patients with ITP.
2. Methods
Human samples
The Vice Chancellor for Research approved this study. Peripheral blood was obtained from patients with acute and chronic ITP (defined as passed >1 year since diagnosis) and closely age-matched normal healthy volunteers (often from patients’ relatives) with their informed consent. In practice, we did not find chronic ITP except for 3 cases. Therefore, our study is based on patients with acute type of disease. The control group consisted of people who had no history of diseases and were often selected from the family of the patients.
The inclusion criteria were lacking Intravenous Immunoglobulin (IVIg) in the past 6 months; not having steroid therapy for at least 2 months; not reporting any previous recurrent infections, documented primary or secondary immune deficiency (due to known diseases), autoimmune disorders, or other acute or chronic disorders. Then, every patient diagnosed with ITP entered the study. The patients were between 6 months and 18 years old. We evaluated age, gender, and blood indexing which included WBC, Hb, platelet count, previous treatments, age of diagnosis, serum immunoglobulins (IgG, IgE, IgM, IgA, IgG1, IgG2, IgG3, IgG4) by nephelometry, antistreptolysin O (ASO) titer by agglutination, CH50, C3, C4 by nephelometry, CD19 by flow cytometry, and anti-tetanus antibody titer by ELISA.
Acute ITP is defined as thrombocytopenia (<150000 platelets) for a duration of up to one year. Chronic ITP refers to thrombocytopenia (<150000 platelets) for more than one year. The sample size selection was based on previous studies. All data were gathered from a questionnaire form. A sample of 7 mL of the patient`s blood was taken and sent to the laboratory for analysis. We used Minineph (Bingham, UK) kits for immunoglobulins and complements. Besides, we used Bionik company kits from Canada for ASO titer and CD19 flow cytometry. ELISA IDL kits from Italy was used for anti-tetanus antibody titer. All patients and healthy people had consented to participate in this study.
According to the following formula and based on Ohali et al. study (that the complement system may indicate a possible role in ITP patients), we select 25 people for each group (10).
Immune Thrombocytopenia (ITP) is an immune and or autoimmune disease with bleeding tendency due to accelerated platelet destruction as well as decreased platelet production mediated in part by autoantibodies. ITP has several etiologies, natural histories, and responses to treatment. In children with ITP, there is sometimes a history of recent infections. In the endemic areas, in adults with ITP, Helicobacter pylori, human immune deficiency virus, or hepatitis C are implicated (1). Vaccination can also induce ITP, such as the measles-mumps-rubella vaccine (MMR, ~1:40000). Some cases of ITP due to vaccination (hepatitis B, diphtheria-tetanus-pertussis, and hepatitis A vaccines) have been reported in children (2).
In patients with ITP, the platelet-autoreactive T cells induce interleukin-2 (IL-2) and interferon-gamma (IFN-γ) predominantly, then with alteration in regulatory T cells (Treg) number and function, a shift toward stimulatory monocytes with enhanced Fcγ receptors (FcγR)-mediated phagocytic capacity causes immune dysregulation in ITP. Some researchers reported increased Th17 cells or IL-17 cytokine in ITP patients whereas other reports did not detect any differences. Resolution in Treg function in ITP patients induces increased platelet counts after the use of these agents (3). Similar results have been reported with rituximab that improve Treg function. High-dose dexamethasone treatment increases the number of circulating Treg cells and decreases Th1 cells (4, 5).
Immunodeficiency disorders are diseases of defective innate adaptive immunity with diverse clinical phenotypes and genotypes manifested by recurrent severe infections, autoimmunity, and lymphoproliferation (6). Antibody deficiencies are more common in primary immune deficiency followed by well-defined syndromes with immunodeficiency, phagocytic disorders, complement deficiencies, immunodeficiencies affecting cellular and humoral immunity in nine patients, disease of immune dysregulation and defects in intrinsic and innate immunity (7).
In primary immune deficiency, especially in antibody disorders, chronic and recurrent infections, chronic inflammation, and autoimmunity are more common. Besides, they tend to develop malignancies and lymphoproliferative disorders. Prevalence of autoimmune disorders in Common Variable Immunodeficiency (CVID), hyper IgM (HIgM), secretory IgA deficiency (SIgAD) and X-linked agammaglobulinemia (XLA) patients were 38 (30.4%), 13 (40.6%), 14 (22.2%), and 11 (18.3%), respectively. Prevalence of ITP in CVID, HIgM, SIgAD, and XLA patients were 5 (4%), 5 (15.5%), 0 (0%), and 3 (5%), respectively (8, 9). In the present study, we explored the role of humoral and complement systems in patients with ITP.
2. Methods
Human samples
The Vice Chancellor for Research approved this study. Peripheral blood was obtained from patients with acute and chronic ITP (defined as passed >1 year since diagnosis) and closely age-matched normal healthy volunteers (often from patients’ relatives) with their informed consent. In practice, we did not find chronic ITP except for 3 cases. Therefore, our study is based on patients with acute type of disease. The control group consisted of people who had no history of diseases and were often selected from the family of the patients.
The inclusion criteria were lacking Intravenous Immunoglobulin (IVIg) in the past 6 months; not having steroid therapy for at least 2 months; not reporting any previous recurrent infections, documented primary or secondary immune deficiency (due to known diseases), autoimmune disorders, or other acute or chronic disorders. Then, every patient diagnosed with ITP entered the study. The patients were between 6 months and 18 years old. We evaluated age, gender, and blood indexing which included WBC, Hb, platelet count, previous treatments, age of diagnosis, serum immunoglobulins (IgG, IgE, IgM, IgA, IgG1, IgG2, IgG3, IgG4) by nephelometry, antistreptolysin O (ASO) titer by agglutination, CH50, C3, C4 by nephelometry, CD19 by flow cytometry, and anti-tetanus antibody titer by ELISA.
Acute ITP is defined as thrombocytopenia (<150000 platelets) for a duration of up to one year. Chronic ITP refers to thrombocytopenia (<150000 platelets) for more than one year. The sample size selection was based on previous studies. All data were gathered from a questionnaire form. A sample of 7 mL of the patient`s blood was taken and sent to the laboratory for analysis. We used Minineph (Bingham, UK) kits for immunoglobulins and complements. Besides, we used Bionik company kits from Canada for ASO titer and CD19 flow cytometry. ELISA IDL kits from Italy was used for anti-tetanus antibody titer. All patients and healthy people had consented to participate in this study.
According to the following formula and based on Ohali et al. study (that the complement system may indicate a possible role in ITP patients), we select 25 people for each group (10).
Statistical analysis
After performing the Kolmogorov-Smirnov test and verifying the normal distribution of variables, quantitative variables were presented with mean and standard deviation and qualitative variables with frequency. The independent t test evaluated the mean difference of variables. Descriptive statistics and statistical analyses were performed in SPSS 25. In all cases, a two-sided P value less than 0.05 was considered statistically significant.
3. Results
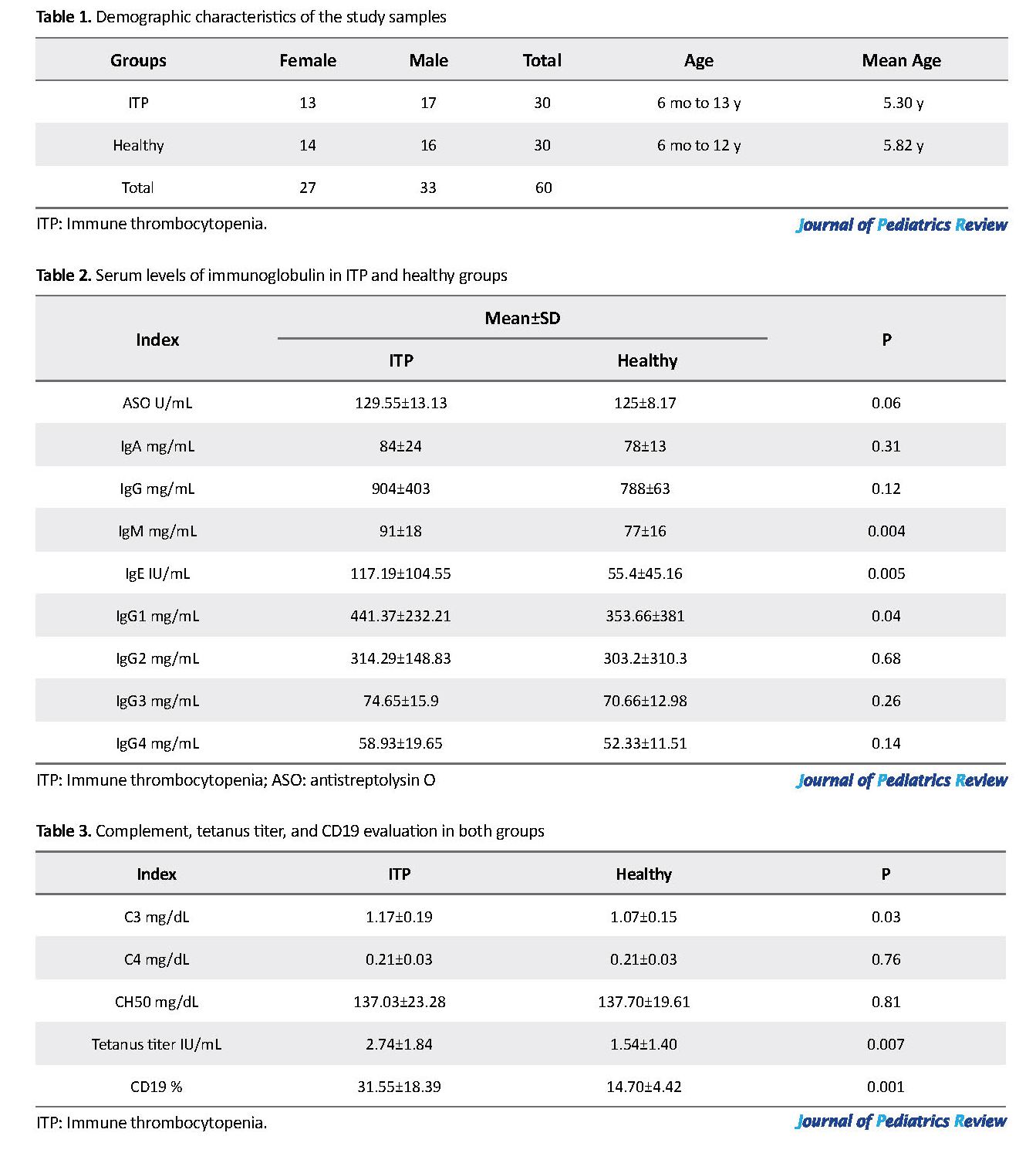
A total of 60 samples were included in our study. Twenty-seven people were female, and 33 were male with an age range between 6 months and 18 years (mean age 5.62 years) (Table 1). Out of our total number of patients, 27 had acute ITP and only 3 had chronic ITP. Clinical symptoms included petechiae and purpura along with other symptoms of ITP, such as abdominal pain or joint pain. Sometimes laboratory tests revealed cases of occult gastrointestinal bleeding. None of the bleedings was evident. All of the acute ITP patients responded to one-time treatment with intravenous immunoglobulin, but chronic cases required frequent treatments. Serum levels of immunoglobulin were within normal range both in the ITP and healthy groups but in the ITP group was higher (Table 2). Table 3 presents the values of complement, tetanus titer and CD19.
After performing the Kolmogorov-Smirnov test and verifying the normal distribution of variables, quantitative variables were presented with mean and standard deviation and qualitative variables with frequency. The independent t test evaluated the mean difference of variables. Descriptive statistics and statistical analyses were performed in SPSS 25. In all cases, a two-sided P value less than 0.05 was considered statistically significant.
3. Results
A total of 60 samples were included in our study. Twenty-seven people were female, and 33 were male with an age range between 6 months and 18 years (mean age 5.62 years) (Table 1). Out of our total number of patients, 27 had acute ITP and only 3 had chronic ITP. Clinical symptoms included petechiae and purpura along with other symptoms of ITP, such as abdominal pain or joint pain. Sometimes laboratory tests revealed cases of occult gastrointestinal bleeding. None of the bleedings was evident. All of the acute ITP patients responded to one-time treatment with intravenous immunoglobulin, but chronic cases required frequent treatments. Serum levels of immunoglobulin were within normal range both in the ITP and healthy groups but in the ITP group was higher (Table 2). Table 3 presents the values of complement, tetanus titer and CD19.
Based on the independent t test results, the differences between 2 groups in the mean serum levels of IgM, IgE, IgG1, C3, tetanus antibody, and CD19 were statistically significant.
4. Discussion
Infections are not common in acute or chronic ITP. However, ITP usually occurs after upper respiratory infections. A few research studies have been conducted on immunodeficiency in ITP patients. In the present research, we evaluated the humoral and complement deficiencies in patients with ITP. Our findings showed that humoral and complement components were normal in all patients. Based on the independent t test results, the mean serum levels of IgM, IgE, IgG1, C3, tetanus antibody, and CD19 were significantly higher in ITP patients. Common variable immunodeficiency (CVID) is a frequent disorder associated with autoimmune diseases (4%-23%) such as ITP, immune hemolytic anemia, and neutropenia. In many cases, autoimmune events are the first manifestations of a primary immune deficiency (9, 11). In another study, ITP was 4.6% prevalent in primary antibody deficiencies, being more common in HIgM disease as compared with the CVID (8).
Similar to our study, other studies showed that CD10, CD19, and CD20 B-cell markers were significantly higher in children with acute and or chronic ITP in bone marrow aspiration (12, 13). CD19 has been bone marrow in the group of ITP patients because it needed to diagnose them. However, in the control group, ethically peripheral blood CD19 performed. In our study, although CD19 was not comparable between the two groups, its percentage in patients was significantly higher. Rahiminejad et al. evaluated the humoral immune system in children and adults (3-51 years old) with chronic ITP. Their patients had no history of recurrent infections similar to our study.
Thirty-eight percent of their chronic ITP patients had signs of primary immunodeficiency. Only 2 patients had selective IgA deficiency (5.0%). There was no difference between patients and controls in any of the immunoglobulin serum levels (IgA, IgG, IgG subclasses, and IgM). Four patients (11.1%) had low IgG subclasses (IgG2 levels) and associated insufficient antibody response to the pneumococcal vaccine (14). This study showed a substantial slight increase in antibody response levels to 23-valent PPV (pneumococcal polysaccharide vaccine) in ITP patients in comparison with the normal responders (14). However, in our study, ITP patients had a higher response to the tetanus vaccine compared with the healthy group (P=0.007). Therefore, unlike Rahiminejad`s research, we did not find Specific Antibody Deficiency (SAD) in ITP patients. Perhaps because our patients, except for three, were of an acute type.
Studies support that peripheral CD19+ subsets based on CD24 and CD38 expression have significantly changed in ITP patients (15, 16). These studies have found impaired IL-10 response in stimulated B cells and a reduced B-cell suppressive activity in ITP patients with low platelet counts (4, 17, 18). Memory B cells deficiency has been reported in patients with ITP. Also, CD16+ blood monocytes can modulate and suppress IL-17+CD4+ and Treg development due to the proliferation of IFN-γ that observed in ITP patients (1, 19). Because of our study limitations, we could not carry out all tests for humoral and complementary systems. Also, the number of chronic ITP patients was low.
5. Conclusions
Humoral and complementary immunodeficiency in our ITP patients is unlikely, perhaps because our patients, except for three, were of an acute type. However, in patients with ITP, checking the immune system without a history of serious or repeated infection is not necessary.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Research vice chancellor of Mazandaran University of Medical Sconces (Code: 2186).
Funding
The Vice Chancellor of Research supported this work. This article is based on a pediatric resident thesis submitted by Fatemeh Mohammadi Kharkeshi done at Mazandaran University of Medical Sciences, Sari, Iran.
Authors contribution
Conceptualization and original idea: Hossein Karamid; Study concept and design and manuscript: Javad Ghaffari; Analysis and interpretation of the data: Siavash Moradi; Technical and material support: Laleh Vahedi; Data gathering: Fatemeh Mohammadi-Kharkeshi contributed to
Conflict of interest
The authors declared no conflict of interest.
Acknowledgements
The authors offer their profound thanks to the patients and their parents and personnel of Oncology Ward for their contribution to this study.
References
Zhong H, Bao W, Li X, Miller A, Seery C, Haq N, et al. CD16+ monocytescontrol T-cell subset development in immune thrombocytopenia. Blood. 2012; 120(16):3326-35. [DOI:10.1182/blood-2012-06-434605] [PMID] [PMCID]
Lazarus AH, Semple JW, Cines DB. Innate and adaptive immunity in ITP. Seminars in hematology. 2013; 50(1):S68-70. [DOI:10.1053/j.seminhematol.2013.03.012] [PMID] [PMCID]
Bao W, Bussel JB, Heck S, He W, Karpoff M, Boulad N, et al. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood. 2010; 116(22):4639-45. [DOI:10.1182/blood-2010-04-281717] [PMID] [PMCID]
Ling Y, Cao X, Yu Z, Ruan C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. European Journal of Haematology. 2007; 79(4):310-6. [DOI:10.1111/j.1600-0609.2007.00917.x] [PMID]
Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood. 2008; 112(4):1147-50. [DOI:10.1182/blood-2007-12-129262] [PMID]
Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. The Journal of Allergy and Clinical Immunology. 2018; 141(4):1450-8. [DOI:10.1016/j.jaci.2017.06.049] [PMID]
Mohammadzadeh I, Moazzami B, Ghaffari J, Aghamohammadi A, Rezaei N. Primary immunodeficiency diseases in Northern Iran. Allergol Immunopathol (Madr). 2017; 45(3):244-50. [DOI:10.1016/j.aller.2016.11.001] [PMID]
Mohammadinejad P, Pourhamdi S, Abolhassani H, Mirminachi B, Havaei A, Masoom SN, et al. Primary antibody deficiency in a tertiary referral hospital: A 30-year experiment. Journal of Investigational Allergology and Clinical Immunology. 2015; 25(6):416-25. [PMID]
Aghamohammadi A, Abolhassani H, Latif A, Tabassomi F, Shokuhfar T, Torabi Sagvand B, et al. Long-term evaluation of a historical cohort of Iranian common variable immunodeficiency patients. Expert Review of Clinical Immunology. 2014; 10(10):1405-17. [DOI:10.1586/1744666X.2014.958469] [PMID]
Ohali M, Maizlish Y, Abramov H, Schlesinger M, Bransky D, Lugassy G. Complement profile in childhood immune thrombocytopenic purpura: A prospective pilot study. Annals of Hematology. 2005; 84(12):812-5. [DOI:10.1007/s00277-005-1085-6] [PMID]
Aghamohammadi A, Farhoudi A, Moin M, Rezaei N, Kouhi A, Pourpak Z, et al. Clinical and immunological features of 65 Iranian patients with common variable immunodeficiency. Clinical and Vaccine Immunology. 2005; 12(7):825-32. [DOI:10.1128/CDLI.12.7.825-832.2005] [PMID] [PMCID]
Alavi S, Aryan Z, Ghazizadeh F, Arabi N, Nikougoftar M, Ebadi M. The immunophenotype of bone marrow lymphocytes in children with immune thrombocytopenic purpura. Pediatric Hematology and Oncology. 2014; 31(6):548-54. [DOI:10.3109/08880018.2014.927944] [PMID]
Guiziry DE, El GW, Farahat N, Hassab H. Phenotypic analysis of bone marrow lymphocytes from children with acute thrombocytopenic purpura. Egyptian Journal of Immunology. 2005; 12(1):9-14. [PMID]
Rahiminejad MS, Mirmohammad Sadeghi M, Mohammadinejad P, Sadeghi B, Abolhassani H, Dehghani Firoozabadi MM, et al. Evaluation of humoral immune function in patients with chronic idiopathic thrombocytopenic purpura. Iranian Journal of Allergy, Asthma and Immunolog. 2013; 12(1):50-6. [DOI:012.01/ijaai.5056] [PMID]
Li X, Zhong H, Bao W, Boulad N, Evangelista J, Haider MA, et al. Defective regulatory B-cell compartment in patients with immune thrombocytopenia. Blood. 2012; 120(16):3318-25. [DOI:10.1182/blood-2012-05-432575] [PMID] [PMCID]
Sakakura M, Wada H, Tawara I, Nobori T, Sugiyama T, Sagawa N, et al. Reduced CD4+CD25+ T cells in patients with idiopathic thrombocytopenic purpura. Thrombosis Research. 2007; 120(2):187-93. [DOI:10.1016/j.thromres.2006.09.008] [PMID]
Liu B, Zhao H, Poon MC, Han Z, Gu D, Xu M, et al. Abnormality of CD4(+) CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. European Journal of Haematology. 2007; 789(2):139-43. [DOI:10.1111/j.1600-0609.2006.00780.x] [PMID]
Yu J, Heck S, Patel V, Levan J, Yu Y, Bussel JB, et al. Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura. Blood. 2008; 112(4):1325-8. [DOI:10.1182/blood-2008-01-135335] [PMID] [PMCID]
Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27(+) IgM(-) IgD(-)) in subgroups of patients with common variable immunodeficiency: A new approach to classify a heterogeneous disease. Blood. 2002; 99(5):1544-51. [DOI:10.1182/blood.V99.5.1544] [PMID]
4. Discussion
Infections are not common in acute or chronic ITP. However, ITP usually occurs after upper respiratory infections. A few research studies have been conducted on immunodeficiency in ITP patients. In the present research, we evaluated the humoral and complement deficiencies in patients with ITP. Our findings showed that humoral and complement components were normal in all patients. Based on the independent t test results, the mean serum levels of IgM, IgE, IgG1, C3, tetanus antibody, and CD19 were significantly higher in ITP patients. Common variable immunodeficiency (CVID) is a frequent disorder associated with autoimmune diseases (4%-23%) such as ITP, immune hemolytic anemia, and neutropenia. In many cases, autoimmune events are the first manifestations of a primary immune deficiency (9, 11). In another study, ITP was 4.6% prevalent in primary antibody deficiencies, being more common in HIgM disease as compared with the CVID (8).
Similar to our study, other studies showed that CD10, CD19, and CD20 B-cell markers were significantly higher in children with acute and or chronic ITP in bone marrow aspiration (12, 13). CD19 has been bone marrow in the group of ITP patients because it needed to diagnose them. However, in the control group, ethically peripheral blood CD19 performed. In our study, although CD19 was not comparable between the two groups, its percentage in patients was significantly higher. Rahiminejad et al. evaluated the humoral immune system in children and adults (3-51 years old) with chronic ITP. Their patients had no history of recurrent infections similar to our study.
Thirty-eight percent of their chronic ITP patients had signs of primary immunodeficiency. Only 2 patients had selective IgA deficiency (5.0%). There was no difference between patients and controls in any of the immunoglobulin serum levels (IgA, IgG, IgG subclasses, and IgM). Four patients (11.1%) had low IgG subclasses (IgG2 levels) and associated insufficient antibody response to the pneumococcal vaccine (14). This study showed a substantial slight increase in antibody response levels to 23-valent PPV (pneumococcal polysaccharide vaccine) in ITP patients in comparison with the normal responders (14). However, in our study, ITP patients had a higher response to the tetanus vaccine compared with the healthy group (P=0.007). Therefore, unlike Rahiminejad`s research, we did not find Specific Antibody Deficiency (SAD) in ITP patients. Perhaps because our patients, except for three, were of an acute type.
Studies support that peripheral CD19+ subsets based on CD24 and CD38 expression have significantly changed in ITP patients (15, 16). These studies have found impaired IL-10 response in stimulated B cells and a reduced B-cell suppressive activity in ITP patients with low platelet counts (4, 17, 18). Memory B cells deficiency has been reported in patients with ITP. Also, CD16+ blood monocytes can modulate and suppress IL-17+CD4+ and Treg development due to the proliferation of IFN-γ that observed in ITP patients (1, 19). Because of our study limitations, we could not carry out all tests for humoral and complementary systems. Also, the number of chronic ITP patients was low.
5. Conclusions
Humoral and complementary immunodeficiency in our ITP patients is unlikely, perhaps because our patients, except for three, were of an acute type. However, in patients with ITP, checking the immune system without a history of serious or repeated infection is not necessary.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Research vice chancellor of Mazandaran University of Medical Sconces (Code: 2186).
Funding
The Vice Chancellor of Research supported this work. This article is based on a pediatric resident thesis submitted by Fatemeh Mohammadi Kharkeshi done at Mazandaran University of Medical Sciences, Sari, Iran.
Authors contribution
Conceptualization and original idea: Hossein Karamid; Study concept and design and manuscript: Javad Ghaffari; Analysis and interpretation of the data: Siavash Moradi; Technical and material support: Laleh Vahedi; Data gathering: Fatemeh Mohammadi-Kharkeshi contributed to
Conflict of interest
The authors declared no conflict of interest.
Acknowledgements
The authors offer their profound thanks to the patients and their parents and personnel of Oncology Ward for their contribution to this study.
References
Zhong H, Bao W, Li X, Miller A, Seery C, Haq N, et al. CD16+ monocytescontrol T-cell subset development in immune thrombocytopenia. Blood. 2012; 120(16):3326-35. [DOI:10.1182/blood-2012-06-434605] [PMID] [PMCID]
Lazarus AH, Semple JW, Cines DB. Innate and adaptive immunity in ITP. Seminars in hematology. 2013; 50(1):S68-70. [DOI:10.1053/j.seminhematol.2013.03.012] [PMID] [PMCID]
Bao W, Bussel JB, Heck S, He W, Karpoff M, Boulad N, et al. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood. 2010; 116(22):4639-45. [DOI:10.1182/blood-2010-04-281717] [PMID] [PMCID]
Ling Y, Cao X, Yu Z, Ruan C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. European Journal of Haematology. 2007; 79(4):310-6. [DOI:10.1111/j.1600-0609.2007.00917.x] [PMID]
Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood. 2008; 112(4):1147-50. [DOI:10.1182/blood-2007-12-129262] [PMID]
Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. The Journal of Allergy and Clinical Immunology. 2018; 141(4):1450-8. [DOI:10.1016/j.jaci.2017.06.049] [PMID]
Mohammadzadeh I, Moazzami B, Ghaffari J, Aghamohammadi A, Rezaei N. Primary immunodeficiency diseases in Northern Iran. Allergol Immunopathol (Madr). 2017; 45(3):244-50. [DOI:10.1016/j.aller.2016.11.001] [PMID]
Mohammadinejad P, Pourhamdi S, Abolhassani H, Mirminachi B, Havaei A, Masoom SN, et al. Primary antibody deficiency in a tertiary referral hospital: A 30-year experiment. Journal of Investigational Allergology and Clinical Immunology. 2015; 25(6):416-25. [PMID]
Aghamohammadi A, Abolhassani H, Latif A, Tabassomi F, Shokuhfar T, Torabi Sagvand B, et al. Long-term evaluation of a historical cohort of Iranian common variable immunodeficiency patients. Expert Review of Clinical Immunology. 2014; 10(10):1405-17. [DOI:10.1586/1744666X.2014.958469] [PMID]
Ohali M, Maizlish Y, Abramov H, Schlesinger M, Bransky D, Lugassy G. Complement profile in childhood immune thrombocytopenic purpura: A prospective pilot study. Annals of Hematology. 2005; 84(12):812-5. [DOI:10.1007/s00277-005-1085-6] [PMID]
Aghamohammadi A, Farhoudi A, Moin M, Rezaei N, Kouhi A, Pourpak Z, et al. Clinical and immunological features of 65 Iranian patients with common variable immunodeficiency. Clinical and Vaccine Immunology. 2005; 12(7):825-32. [DOI:10.1128/CDLI.12.7.825-832.2005] [PMID] [PMCID]
Alavi S, Aryan Z, Ghazizadeh F, Arabi N, Nikougoftar M, Ebadi M. The immunophenotype of bone marrow lymphocytes in children with immune thrombocytopenic purpura. Pediatric Hematology and Oncology. 2014; 31(6):548-54. [DOI:10.3109/08880018.2014.927944] [PMID]
Guiziry DE, El GW, Farahat N, Hassab H. Phenotypic analysis of bone marrow lymphocytes from children with acute thrombocytopenic purpura. Egyptian Journal of Immunology. 2005; 12(1):9-14. [PMID]
Rahiminejad MS, Mirmohammad Sadeghi M, Mohammadinejad P, Sadeghi B, Abolhassani H, Dehghani Firoozabadi MM, et al. Evaluation of humoral immune function in patients with chronic idiopathic thrombocytopenic purpura. Iranian Journal of Allergy, Asthma and Immunolog. 2013; 12(1):50-6. [DOI:012.01/ijaai.5056] [PMID]
Li X, Zhong H, Bao W, Boulad N, Evangelista J, Haider MA, et al. Defective regulatory B-cell compartment in patients with immune thrombocytopenia. Blood. 2012; 120(16):3318-25. [DOI:10.1182/blood-2012-05-432575] [PMID] [PMCID]
Sakakura M, Wada H, Tawara I, Nobori T, Sugiyama T, Sagawa N, et al. Reduced CD4+CD25+ T cells in patients with idiopathic thrombocytopenic purpura. Thrombosis Research. 2007; 120(2):187-93. [DOI:10.1016/j.thromres.2006.09.008] [PMID]
Liu B, Zhao H, Poon MC, Han Z, Gu D, Xu M, et al. Abnormality of CD4(+) CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. European Journal of Haematology. 2007; 789(2):139-43. [DOI:10.1111/j.1600-0609.2006.00780.x] [PMID]
Yu J, Heck S, Patel V, Levan J, Yu Y, Bussel JB, et al. Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura. Blood. 2008; 112(4):1325-8. [DOI:10.1182/blood-2008-01-135335] [PMID] [PMCID]
Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27(+) IgM(-) IgD(-)) in subgroups of patients with common variable immunodeficiency: A new approach to classify a heterogeneous disease. Blood. 2002; 99(5):1544-51. [DOI:10.1182/blood.V99.5.1544] [PMID]
Type of Study: Original Article |
Subject:
Allergy and Clinical Immunology
Received: 2019/04/7 | Accepted: 2019/06/9 | Published: 2020/04/1
Received: 2019/04/7 | Accepted: 2019/06/9 | Published: 2020/04/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
