
Tue, Jun 9, 2026


Volume 13, Issue 2 (4-2025)
J. Pediatr. Rev 2025, 13(2): 133-138 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Ghorbani-khosroshahi N, Azad G, Nili F, Mohkam M, Gholamali-Majdabadi H. Atypical Presentation of Thin Basement Membrane Disease in a Child: Diagnostic and Management Considerations. J. Pediatr. Rev 2025; 13 (2) :133-138
URL: http://jpr.mazums.ac.ir/article-1-729-en.html
URL: http://jpr.mazums.ac.ir/article-1-729-en.html
Neda Ghorbani-khosroshahi1 

 , Ghazal Azad *2 , Fatemeh Nili3 , Masoumeh Mohkam1 , Hananeh Gholamali-Majdabadi1
, Ghazal Azad *2 , Fatemeh Nili3 , Masoumeh Mohkam1 , Hananeh Gholamali-Majdabadi1
, Ghazal Azad *2 , Fatemeh Nili3 , Masoumeh Mohkam1 , Hananeh Gholamali-Majdabadi1
1- Pediatric Nephrology Research Center, Research Institute for Children’s Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2- School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran. ,ghazalazad79@gmail.com
3- Department of Pathology, Cancer Institute, Imam Khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran, Iran.
2- School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran. ,
3- Department of Pathology, Cancer Institute, Imam Khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran, Iran.
Keywords: Electron microscopy, Hematuria, Glomerular basement membrane (GBM), Henoch–Schoenlein Purpura (immunoglobulin A [IgA] vasculitis), Renal biopsy
Full-Text [PDF 1932 kb]
(841 Downloads)
| Abstract (HTML) (2112 Views)
Full-Text: (291 Views)
Introduction
Tea-colored urine in cases of macroscopic hematuria often indicates a glomerular origin, with differential diagnoses, commonly including thin basement membrane disease (TBMD) and Henoch-Schoenlein Purpura (HSP) [1]. TBMD is the most prevalent cause of persistent glomerular hematuria in children and adults, affecting at least 1% of the population [2]. Patients with TBMD frequently present with hematuria and or mild proteinuria, typically while maintaining normal renal function [3]. HSP is a multisystemic vasculitis characterized by its hallmark rash and potential involvement of the gastrointestinal tract and kidneys, manifesting as acute abdominal pain and mild microscopic urinary abnormalities [4]. Conversely, TBMD is characterized by recurrent episodes of hematuria, often precipitated by infections or physical exertion [5].
The primary overlap between HSP and TBMD lies in renal involvement, as extrarenal symptoms are generally absent in TBMD. Hematuria is a common feature of these conditions; however, in HSP, it is usually accompanied by proteinuria and sometimes red blood cell (RBC) casts. In contrast, hematuria in TBMD is typically not associated with significant proteinuria. Additionally, in children, loin pain, which is occasionally observed in TBMD [2], may be misinterpreted as abdominal pain, a classic symptom of HSP. Despite these similarities, distinguishing between the two conditions is generally straightforward. In HSP, characteristic rashes and immunoglobulin A (IgA) deposits in the mesangium on kidney biopsy provide critical diagnostic clues. Conversely, TBMD is identified by uniform thinning of the glomerular basement membrane (GBM) on biopsy. A positive family history of hematuria, frequently present in TBMD but uncommon in HSP, further aids in differentiation.
The primary concerns associated with TBMD are the misunderstanding and anxiety surrounding the diagnosis, as well as the risks and unnecessary costs related to unwarranted investigations. However, a subset of patients with TBMD may progress to renal impairment, although the underlying mechanisms remain unclear beyond known predisposing factors, such as hypertension and proteinuria [2]. Therefore, this study reviews this diagnostic pathway and describes the atypical manifestations of TBMD to prevent potential misdiagnoses.
Case Presentation
A 4-year-old boy was admitted to our hospital with complaints of severe abdominal pain and vomiting, without any history of abdominal trauma. He was diagnosed with appendicitis and underwent an appendectomy; however, according to the biopsy results, no acute inflammation was observed, and only reactive lymphoid follicular hyperplasia was noted. Five days later, the patient returned with recurrent abdominal pain and vomiting. As surgical evaluations were unremarkable, he was referred to the Gastroenterology Department, where an endoscopy suggested mild chronic inflammation in the esophagus, mild chronic inactive gastritis, mild-moderate chronic inflammation in the duodenum, focal mild increased intraepithelial lymphocytes, and no evidence of Helicobacter pylori. During the hospitalization, the patient’s mother noted a change in urine color. Urinalysis showed RBC=50-60/HPF, white blood cells (WBC)=30-40/HPF, specific gravity (SG)=1.025, blood=2+, protein=3+, dysmorphic RBC=7% (normal range: Under 20%). A 24-h urine test was also performed, and its results were as follows: Urine volume=800 mL, protein=1488 mg, creatinine=258 mg/day, and protein (mg/kg)=106.2 mg/kg. A nephrology consultation was requested for the patient with this clinical presentation, and the patient was subsequently referred to the Nephrology Department. Shortly thereafter, he developed non-blanching red maculopapular skin lesions on both lower legs, with a history of arthralgia in recent months; based on this complaint, a rheumatology consultation was requested for the patient. The rheumatologists stated that given the combination of hematuria, purpura on both legs, abdominal pain, and history of arthralgia, a differential diagnosis of HSP or IgA vasculitis was considered. Additionally, due to the presence of proteinuria and hematuria, other etiologies of glomerulonephritis were also suspected. Accordingly, additional serological, immunological, viral, and coagulation tests were performed, all of which returned normal results (anti–double-stranded DNA antibody=8.15, antinuclear antibody <1/40, complement component 4=40 [normal range 9–36], total hemolytic complement [CH50]=112 [normal range 51–150], international normalized ratio=1, prothrombin time=12 s, partial thromboplastin time=30 s, white blood cell count=17800/µL, hemoglobin=11 g/dL, and platelet count=705000/µL). Renal ultrasound revealed a right kidney measuring 82 mm and a left kidney measuring 80 mm, which is larger than the normal range for the patient’s age.
The patient had a medical history of allergic rhinitis, with no family history of hematuria or kidney disease. His only prior surgery was an appendectomy. On admission, he weighed 14 kg, and his vital signs, including blood pressure, were within normal limits. Physical examination was unremarkable except for maculopapular rashes on the lower limbs and occasional arthralgia.
Treatment and follow-up
After a week, due to the lack of response to the 2 mg/kg/day oral prednisolone dose for proteinuria, the patient received three 15 mg/kg/day doses of pulse corticosteroids over three days because of the continuous of severe proteinuria (the results of the 24-h urine test before starting the pulse corticosteroid treatment: Urine volume=500 mL, protein=4455 mg, creatinine=260 mg/day, protein (mg/kg)=318.2 mg/kg). Since this treatment was effective in reducing the patient’s proteinuria, he was discharged with a prescription for enalapril (0.2 mg/kg/day) and prednisolone (2 mg/kg/day).
Approximately 60 days later, following a tapering in the corticosteroid dose and after the patient experienced a common cold, hematuria reoccurred, as indicated by the urine analysis (RBC=many, protein=2+, blood=3+). Based on recurrent episodes of hematuria, proteinuria, and lower and upper respiratory tract infections, the possibility of lupus or anti-neutrophil cytoplasmic antibody–associated vasculitis (ANCA)-associated vasculitis was considered for the patient. Therefore, additional tests were ordered, yielding normal results: C3=1.42, C4=0.35, CH50=0.87, cytoplasmic ANCA=0.15, perinuclear ANCA=0.12, blood urea nitrogen=15.4, creatinine=0.61. The normal level of C3 supported the likelihood of HSP and TBMD and ruled out the diagnosis of diseases such as lupus or ANCA-associated vasculitis. The indication for biopsy was made for the patient at this time, but due to the lack of consent from the parents, the patient was discharged with enalapril (0.2 mg/kg/day) and prednisolone (2 mg/kg/day).
Urine analysis was performed during follow-up visits whenever urine discoloration was reported, particularly after upper respiratory infections. The results consistently indicated hematuria with proteinuria: RBC=20-25/HPF, WBC=3-5/HPF, SG=1.013, blood=2+, protein=2+. At this time, given the patient’s clinical presentation, particularly recurrent hematuria triggered by upper respiratory infections, IgA nephropathy was also considered apart from IgA vasculitis. In the intervals between infections, when there was no complaint of urine discoloration, the patient’s urinalysis results were nearly normal.
Finally, 23 months after the patient’s initial hospitalization, a renal biopsy was performed due to persistent recurrent hematuria, especially following upper respiratory infections and episodes of sinusitis lasting 3–5 days.
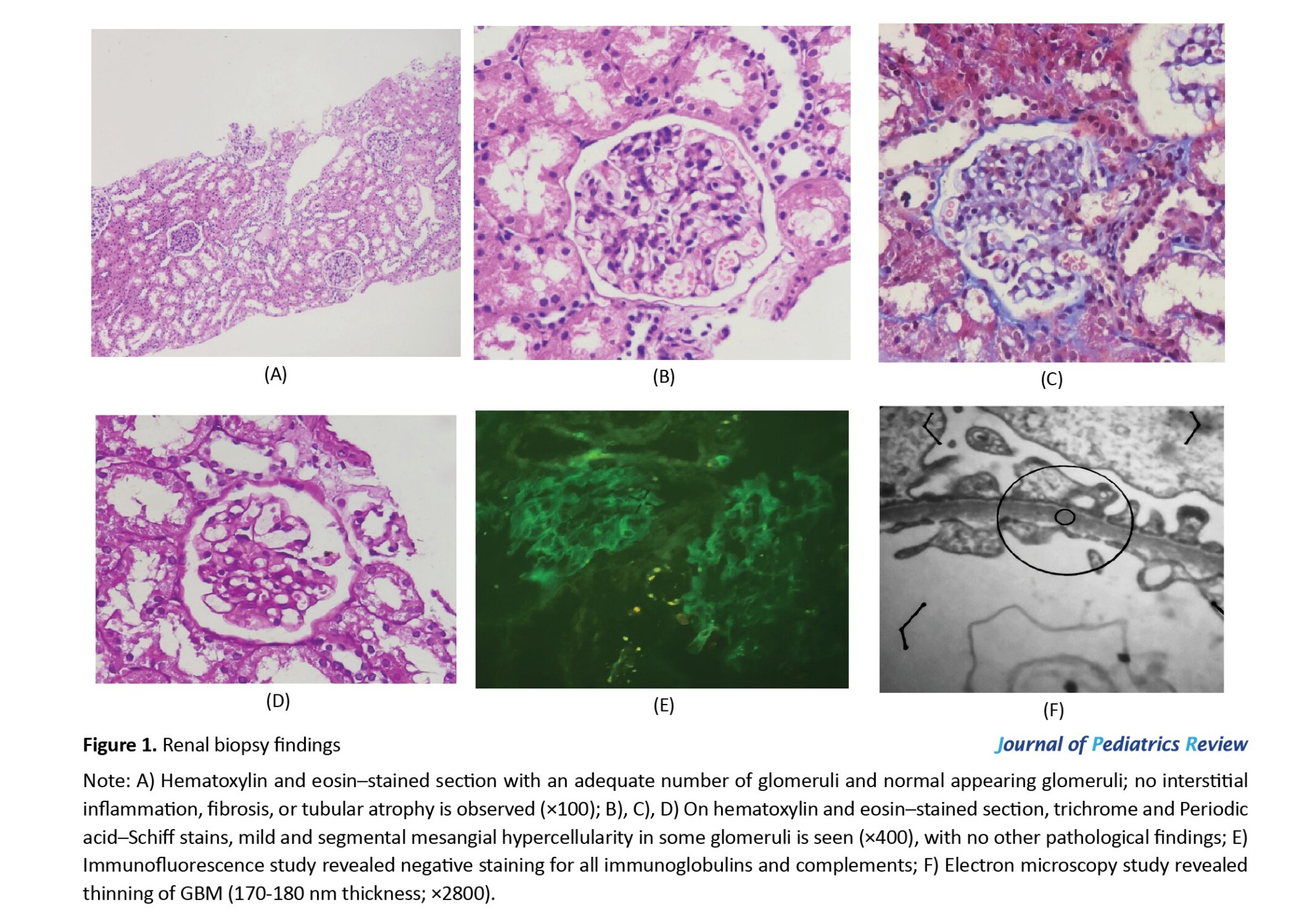
Based on the pathological results, the light microscopy findings were nearly normal, with no evidence of global or segmental sclerosis in the glomeruli (Figures 1A, 1B, 1C and 1D). The immunofluorescence results were negative (Figure 1E), and the electron microscopy study demonstrated global thinning of the GBM with an average thickness of 170 nm (Figures 1F). As the GBM thickness was at the lower limit of normal, a diagnosis of TBMD was established. Since the results of the biopsy, including electron microscopy and immunofluorescence studies, especially the absence of IgA sediment, ruled out the diagnosis of HSP in this case, the patient’s prednisolone and enalapril were tapered and eventually discontinued.
Discussion
We diagnosed a patient with TBMD based on biopsy findings. The patient presented with recurrent macroscopic hematuria triggered by upper respiratory infection or sinusitis, accompanied by atypical manifestations such as purpura-like lesions, nausea, vomiting, and abdominal pain. Urinalysis revealed hematuria with mild to moderate proteinuria. Despite these atypical presentations that resembled IgA vasculitis, the characteristic feature of TBMD is intermittent asymptomatic microscopic hematuria [6]. Nonetheless, the patient’s normal renal function and blood pressure supported the diagnosis.
TBMD is the leading cause of persistent glomerular bleeding in both children and adults, affecting at least 1% of the population. Individuals with TBMD typically experience hematuria, minimal proteinuria, normal kidney function, a consistently thinned GBM, and often have a family history of hematuria. The condition generally follows a benign clinical course [2]. In most cases, TBMD results from a defect in type IV collagen, a key structural component of GBM. This disorder arises from mutations in the COL4A3/COL4A4 genes, which are responsible for producing the α3-α4 chains of collagen [5]. The GBM is delicate and prone to damage by proteases, leading to temporary, localized ruptures of the membrane. These ruptures in the glomerular filtration barrier enable RBCs to leak from the capillary space into the Bowman space of the glomerulus that resulting in hematuria [7, 8]. Approximately two-thirds of individuals with TBMD have at least one affected family member with hematuria when five relatives are screened, following an autosomal dominant inheritance pattern. However, one-third of individuals with TBMD have no family history of hematuria. This can be attributed to genetic factors such as de novo mutations or incomplete penetrance, which may explain the absence of a positive family history in this patient [2].
The initial diagnostic evaluations of patients with hematuria include urinalysis, microscopic examination of urinary sediment, and laboratory assessment of renal function. In our case, urine analysis revealed hematuria accompanied by proteinuria. In patients with TBMD, if proteinuria is detected on dipstick testing, it should be quantified using the urine protein-to-creatinine ratio [9]. Renal biopsy remains the definitive diagnostic method for TBMD and was warranted in this case due to the presence of atypical and extra-renal manifestations, including purpura-like lesions, recurrent macroscopic hematuria, and proteinuria. The light microscopic appearance of the renal biopsy in TBMD is nearly normal, with only mild mesangial cell proliferation and slight matrix expansion [2]. Immunofluorescence findings are typically negative. Electron microscopy is the most important part that reveals a uniform thinning of the GBM. Additionally, GBM thickness varies with age, gender, methods of tissue preparation, and measurement method. However, the World Health Organization (WHO) has proposed a diagnostic threshold of 180 nm for children aged 2 to 11 years [5]. In this patient, pathological findings demonstrated a global thinning of 170 mm of GBM with an almost normal appearance under light microscopy. The combined use of electron microscopy and immunohistochemical analysis enhances both the sensitivity and specificity for diagnosing TBMD [5].
As previously mentioned, the main presentation of this patient was recurrent macroscopic hematuria, particularly after every URI. While microscopic hematuria is a hallmark of TBMD, macroscopic hematuria occurs in only 34% of pediatric TBMD cases, typically following exercise or infection [2]. Gross hematuria can sometimes be accompanied by flank pain [5]; however, this has not been reported in pediatric cases [2], including in our patient. Proteinuria is observed in only 6% of children with TBMD and is usually ≤500 mg/day [2], which does not align with the levels seen in our patient. Notably, significant proteinuria in TBMD may result from secondary glomerulosclerosis or other superimposed glomerular lesions [10], though no evidence of sclerosis was found in this patient’s renal biopsy. TBMD is not associated with extra-renal manifestations and does not present with high-tone sensorineural hearing loss, anterior lenticonus, or dot-and-fleck retinopathy, which are characteristic of Alport syndrome, another inherited disorder affecting the GBM. Although hypercalciuria, hyperuricosuria, and nephrolithiasis have been reported in TBMD, these associations remain unconfirmed [11], and our patient exhibited none of these features. The presence of maculopapular rashes, alongside hematuria, initially raised suspicion for HSP or IgA vasculitis; however, the recurrent disease course and frequent relapses were inconsistent with that diagnosis. Therefore, to reach a diagnosis, we performed a biopsy, and the negative immunofluorescence findings regarding IgA deposition were against the diagnosis of HSP. Most individuals with TBMD maintain normal renal function, but proteinuria and hypertension are key risk factors for potential renal impairment [12].
One of the most critical differential diagnoses for TBMD is IgA nephropathy, which occurs more frequently in males and is typically characterized by fluctuating urinary RBC counts, manifesting as macroscopic hematuria during infections and microscopic hematuria between episodes. IgA nephropathy is also commonly associated with proteinuria exceeding 500 mg/day and progressive renal impairment, whereas TBMD typically does not present with such progression. Furthermore, unlike TBMD, IgA nephropathy is not typically associated with a positive family history of hematuria [2]. In our case, the recurrence of hematuria following each URI and the absence of a family history were suggestive of IgA nephropathy. Henoch-Schoenlein, also known as IgA vasculitis, and IgA nephropathy are at two ends of a spectrum of diseases that were considered during the diagnostic process. The absence of characteristic Henoch-Schoenlein skin rashes during the relapses of the disease in this case, along with the recurrence of hematuria following each cold, shifted our diagnosis from IgA vasculitis to IgA nephropathy. Interestingly, studies have reported IgA nephropathy coexisting with TBMD in 2% to 39% of cases, suggesting a higher-than-random association. However, patients with both TBMD and IgA nephropathy do not appear to have a higher risk of proteinuria, hypertension, or renal impairment compared to those with isolated IgA nephropathy [2].
Currently, there is no specific, evidence-based management strategy for TBMD [13]. Most patients with isolated microscopic hematuria have a benign clinical course and do not require any specific therapy [5]. Once diagnosed, management involves regular clinical monitoring for the onset of hypertension, including routine blood pressure assessments. Laboratory follow-up should include serum creatinine, blood urea nitrogen levels, and urinalyses with urine protein-to-creatinine ratio measurements to monitor renal impairment and proteinuria progression. These routine evaluations can be performed by the primary care provider, with specialist referrals as needed every 1 to 2 years [2].
For our patients’ follow-up, urinalysis was conducted whenever urine discoloration was observed, especially after URI. Additionally, serial ultrasounds were performed to assess kidney size and any abnormal findings. If proteinuria or hypertension develops, in addition to lifestyle modifications, pharmacological therapy, involving the use of the maximum tolerated doses of renin-angiotensin-system inhibitors, such as angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, is recommended [6]. In this case, enalapril was prescribed as an angiotensin-converting enzyme inhibitor due to its superior potency, longer duration of action, and potentially improved safety profile compared to alternatives such as captopril [14].
Conclusion
This case highlights the importance of considering TBMD in pediatric patients presenting with recurrent macroscopic hematuria, particularly following URI. Although TBMD is often benign, it may present with atypical features and can be associated with proteinuria, necessitating careful evaluation. Differentiation from HSP, IgA nephropathy, and other glomerular diseases is crucial to ensure appropriate management. Given its hereditary nature, a comprehensive family history and genetic assessment should be considered. Long-term follow-up is essential for monitoring renal function, proteinuria progression, and managing hypertension, thereby preventing potential complications.
Study limitations
In this study, we did not encounter significant limitations. The only issue was the necessity of obtaining family consent for the kidney biopsy of the child, which, due to sensitivities and the family’s reluctance, was delayed for approximately 20 months, disrupting the patient’s diagnosis and treatment course.
Ethical Considerations
Compliance with ethical guidelines
All procedures performed in studies involving human participants followed the ethical standards of the Ethics Committee of Shahid Beheshti University of Medical Sciences, Tehran, Iran. Informed consent was obtained from the patient described in this case report.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualisation and study design: Masoumeh Mohkam and Neda Ghorbani-Khosroshahi; Data acquisition: Neda Ghorbani-Khosroshahi; Writing the original draft: Ghazal Azad and Neda Ghorbani-Khosroshahi, and Fatemeh Nili; Review and editing: Ghazal Azad, Neda Ghorbani-khosroshahi, and Masoumeh Mohkam; Supervision: Masoumeh Mohkam.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors thank all patients who participated in the study. The authors also would like to express their gratitude to the staff of the Pediatric Nephrology Research Center of Mofid Children's Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
References
Tea-colored urine in cases of macroscopic hematuria often indicates a glomerular origin, with differential diagnoses, commonly including thin basement membrane disease (TBMD) and Henoch-Schoenlein Purpura (HSP) [1]. TBMD is the most prevalent cause of persistent glomerular hematuria in children and adults, affecting at least 1% of the population [2]. Patients with TBMD frequently present with hematuria and or mild proteinuria, typically while maintaining normal renal function [3]. HSP is a multisystemic vasculitis characterized by its hallmark rash and potential involvement of the gastrointestinal tract and kidneys, manifesting as acute abdominal pain and mild microscopic urinary abnormalities [4]. Conversely, TBMD is characterized by recurrent episodes of hematuria, often precipitated by infections or physical exertion [5].
The primary overlap between HSP and TBMD lies in renal involvement, as extrarenal symptoms are generally absent in TBMD. Hematuria is a common feature of these conditions; however, in HSP, it is usually accompanied by proteinuria and sometimes red blood cell (RBC) casts. In contrast, hematuria in TBMD is typically not associated with significant proteinuria. Additionally, in children, loin pain, which is occasionally observed in TBMD [2], may be misinterpreted as abdominal pain, a classic symptom of HSP. Despite these similarities, distinguishing between the two conditions is generally straightforward. In HSP, characteristic rashes and immunoglobulin A (IgA) deposits in the mesangium on kidney biopsy provide critical diagnostic clues. Conversely, TBMD is identified by uniform thinning of the glomerular basement membrane (GBM) on biopsy. A positive family history of hematuria, frequently present in TBMD but uncommon in HSP, further aids in differentiation.
The primary concerns associated with TBMD are the misunderstanding and anxiety surrounding the diagnosis, as well as the risks and unnecessary costs related to unwarranted investigations. However, a subset of patients with TBMD may progress to renal impairment, although the underlying mechanisms remain unclear beyond known predisposing factors, such as hypertension and proteinuria [2]. Therefore, this study reviews this diagnostic pathway and describes the atypical manifestations of TBMD to prevent potential misdiagnoses.
Case Presentation
A 4-year-old boy was admitted to our hospital with complaints of severe abdominal pain and vomiting, without any history of abdominal trauma. He was diagnosed with appendicitis and underwent an appendectomy; however, according to the biopsy results, no acute inflammation was observed, and only reactive lymphoid follicular hyperplasia was noted. Five days later, the patient returned with recurrent abdominal pain and vomiting. As surgical evaluations were unremarkable, he was referred to the Gastroenterology Department, where an endoscopy suggested mild chronic inflammation in the esophagus, mild chronic inactive gastritis, mild-moderate chronic inflammation in the duodenum, focal mild increased intraepithelial lymphocytes, and no evidence of Helicobacter pylori. During the hospitalization, the patient’s mother noted a change in urine color. Urinalysis showed RBC=50-60/HPF, white blood cells (WBC)=30-40/HPF, specific gravity (SG)=1.025, blood=2+, protein=3+, dysmorphic RBC=7% (normal range: Under 20%). A 24-h urine test was also performed, and its results were as follows: Urine volume=800 mL, protein=1488 mg, creatinine=258 mg/day, and protein (mg/kg)=106.2 mg/kg. A nephrology consultation was requested for the patient with this clinical presentation, and the patient was subsequently referred to the Nephrology Department. Shortly thereafter, he developed non-blanching red maculopapular skin lesions on both lower legs, with a history of arthralgia in recent months; based on this complaint, a rheumatology consultation was requested for the patient. The rheumatologists stated that given the combination of hematuria, purpura on both legs, abdominal pain, and history of arthralgia, a differential diagnosis of HSP or IgA vasculitis was considered. Additionally, due to the presence of proteinuria and hematuria, other etiologies of glomerulonephritis were also suspected. Accordingly, additional serological, immunological, viral, and coagulation tests were performed, all of which returned normal results (anti–double-stranded DNA antibody=8.15, antinuclear antibody <1/40, complement component 4=40 [normal range 9–36], total hemolytic complement [CH50]=112 [normal range 51–150], international normalized ratio=1, prothrombin time=12 s, partial thromboplastin time=30 s, white blood cell count=17800/µL, hemoglobin=11 g/dL, and platelet count=705000/µL). Renal ultrasound revealed a right kidney measuring 82 mm and a left kidney measuring 80 mm, which is larger than the normal range for the patient’s age.
The patient had a medical history of allergic rhinitis, with no family history of hematuria or kidney disease. His only prior surgery was an appendectomy. On admission, he weighed 14 kg, and his vital signs, including blood pressure, were within normal limits. Physical examination was unremarkable except for maculopapular rashes on the lower limbs and occasional arthralgia.
Treatment and follow-up
After a week, due to the lack of response to the 2 mg/kg/day oral prednisolone dose for proteinuria, the patient received three 15 mg/kg/day doses of pulse corticosteroids over three days because of the continuous of severe proteinuria (the results of the 24-h urine test before starting the pulse corticosteroid treatment: Urine volume=500 mL, protein=4455 mg, creatinine=260 mg/day, protein (mg/kg)=318.2 mg/kg). Since this treatment was effective in reducing the patient’s proteinuria, he was discharged with a prescription for enalapril (0.2 mg/kg/day) and prednisolone (2 mg/kg/day).
Approximately 60 days later, following a tapering in the corticosteroid dose and after the patient experienced a common cold, hematuria reoccurred, as indicated by the urine analysis (RBC=many, protein=2+, blood=3+). Based on recurrent episodes of hematuria, proteinuria, and lower and upper respiratory tract infections, the possibility of lupus or anti-neutrophil cytoplasmic antibody–associated vasculitis (ANCA)-associated vasculitis was considered for the patient. Therefore, additional tests were ordered, yielding normal results: C3=1.42, C4=0.35, CH50=0.87, cytoplasmic ANCA=0.15, perinuclear ANCA=0.12, blood urea nitrogen=15.4, creatinine=0.61. The normal level of C3 supported the likelihood of HSP and TBMD and ruled out the diagnosis of diseases such as lupus or ANCA-associated vasculitis. The indication for biopsy was made for the patient at this time, but due to the lack of consent from the parents, the patient was discharged with enalapril (0.2 mg/kg/day) and prednisolone (2 mg/kg/day).
Urine analysis was performed during follow-up visits whenever urine discoloration was reported, particularly after upper respiratory infections. The results consistently indicated hematuria with proteinuria: RBC=20-25/HPF, WBC=3-5/HPF, SG=1.013, blood=2+, protein=2+. At this time, given the patient’s clinical presentation, particularly recurrent hematuria triggered by upper respiratory infections, IgA nephropathy was also considered apart from IgA vasculitis. In the intervals between infections, when there was no complaint of urine discoloration, the patient’s urinalysis results were nearly normal.
Finally, 23 months after the patient’s initial hospitalization, a renal biopsy was performed due to persistent recurrent hematuria, especially following upper respiratory infections and episodes of sinusitis lasting 3–5 days.
Based on the pathological results, the light microscopy findings were nearly normal, with no evidence of global or segmental sclerosis in the glomeruli (Figures 1A, 1B, 1C and 1D). The immunofluorescence results were negative (Figure 1E), and the electron microscopy study demonstrated global thinning of the GBM with an average thickness of 170 nm (Figures 1F). As the GBM thickness was at the lower limit of normal, a diagnosis of TBMD was established. Since the results of the biopsy, including electron microscopy and immunofluorescence studies, especially the absence of IgA sediment, ruled out the diagnosis of HSP in this case, the patient’s prednisolone and enalapril were tapered and eventually discontinued.
Discussion
We diagnosed a patient with TBMD based on biopsy findings. The patient presented with recurrent macroscopic hematuria triggered by upper respiratory infection or sinusitis, accompanied by atypical manifestations such as purpura-like lesions, nausea, vomiting, and abdominal pain. Urinalysis revealed hematuria with mild to moderate proteinuria. Despite these atypical presentations that resembled IgA vasculitis, the characteristic feature of TBMD is intermittent asymptomatic microscopic hematuria [6]. Nonetheless, the patient’s normal renal function and blood pressure supported the diagnosis.
TBMD is the leading cause of persistent glomerular bleeding in both children and adults, affecting at least 1% of the population. Individuals with TBMD typically experience hematuria, minimal proteinuria, normal kidney function, a consistently thinned GBM, and often have a family history of hematuria. The condition generally follows a benign clinical course [2]. In most cases, TBMD results from a defect in type IV collagen, a key structural component of GBM. This disorder arises from mutations in the COL4A3/COL4A4 genes, which are responsible for producing the α3-α4 chains of collagen [5]. The GBM is delicate and prone to damage by proteases, leading to temporary, localized ruptures of the membrane. These ruptures in the glomerular filtration barrier enable RBCs to leak from the capillary space into the Bowman space of the glomerulus that resulting in hematuria [7, 8]. Approximately two-thirds of individuals with TBMD have at least one affected family member with hematuria when five relatives are screened, following an autosomal dominant inheritance pattern. However, one-third of individuals with TBMD have no family history of hematuria. This can be attributed to genetic factors such as de novo mutations or incomplete penetrance, which may explain the absence of a positive family history in this patient [2].
The initial diagnostic evaluations of patients with hematuria include urinalysis, microscopic examination of urinary sediment, and laboratory assessment of renal function. In our case, urine analysis revealed hematuria accompanied by proteinuria. In patients with TBMD, if proteinuria is detected on dipstick testing, it should be quantified using the urine protein-to-creatinine ratio [9]. Renal biopsy remains the definitive diagnostic method for TBMD and was warranted in this case due to the presence of atypical and extra-renal manifestations, including purpura-like lesions, recurrent macroscopic hematuria, and proteinuria. The light microscopic appearance of the renal biopsy in TBMD is nearly normal, with only mild mesangial cell proliferation and slight matrix expansion [2]. Immunofluorescence findings are typically negative. Electron microscopy is the most important part that reveals a uniform thinning of the GBM. Additionally, GBM thickness varies with age, gender, methods of tissue preparation, and measurement method. However, the World Health Organization (WHO) has proposed a diagnostic threshold of 180 nm for children aged 2 to 11 years [5]. In this patient, pathological findings demonstrated a global thinning of 170 mm of GBM with an almost normal appearance under light microscopy. The combined use of electron microscopy and immunohistochemical analysis enhances both the sensitivity and specificity for diagnosing TBMD [5].
As previously mentioned, the main presentation of this patient was recurrent macroscopic hematuria, particularly after every URI. While microscopic hematuria is a hallmark of TBMD, macroscopic hematuria occurs in only 34% of pediatric TBMD cases, typically following exercise or infection [2]. Gross hematuria can sometimes be accompanied by flank pain [5]; however, this has not been reported in pediatric cases [2], including in our patient. Proteinuria is observed in only 6% of children with TBMD and is usually ≤500 mg/day [2], which does not align with the levels seen in our patient. Notably, significant proteinuria in TBMD may result from secondary glomerulosclerosis or other superimposed glomerular lesions [10], though no evidence of sclerosis was found in this patient’s renal biopsy. TBMD is not associated with extra-renal manifestations and does not present with high-tone sensorineural hearing loss, anterior lenticonus, or dot-and-fleck retinopathy, which are characteristic of Alport syndrome, another inherited disorder affecting the GBM. Although hypercalciuria, hyperuricosuria, and nephrolithiasis have been reported in TBMD, these associations remain unconfirmed [11], and our patient exhibited none of these features. The presence of maculopapular rashes, alongside hematuria, initially raised suspicion for HSP or IgA vasculitis; however, the recurrent disease course and frequent relapses were inconsistent with that diagnosis. Therefore, to reach a diagnosis, we performed a biopsy, and the negative immunofluorescence findings regarding IgA deposition were against the diagnosis of HSP. Most individuals with TBMD maintain normal renal function, but proteinuria and hypertension are key risk factors for potential renal impairment [12].
One of the most critical differential diagnoses for TBMD is IgA nephropathy, which occurs more frequently in males and is typically characterized by fluctuating urinary RBC counts, manifesting as macroscopic hematuria during infections and microscopic hematuria between episodes. IgA nephropathy is also commonly associated with proteinuria exceeding 500 mg/day and progressive renal impairment, whereas TBMD typically does not present with such progression. Furthermore, unlike TBMD, IgA nephropathy is not typically associated with a positive family history of hematuria [2]. In our case, the recurrence of hematuria following each URI and the absence of a family history were suggestive of IgA nephropathy. Henoch-Schoenlein, also known as IgA vasculitis, and IgA nephropathy are at two ends of a spectrum of diseases that were considered during the diagnostic process. The absence of characteristic Henoch-Schoenlein skin rashes during the relapses of the disease in this case, along with the recurrence of hematuria following each cold, shifted our diagnosis from IgA vasculitis to IgA nephropathy. Interestingly, studies have reported IgA nephropathy coexisting with TBMD in 2% to 39% of cases, suggesting a higher-than-random association. However, patients with both TBMD and IgA nephropathy do not appear to have a higher risk of proteinuria, hypertension, or renal impairment compared to those with isolated IgA nephropathy [2].
Currently, there is no specific, evidence-based management strategy for TBMD [13]. Most patients with isolated microscopic hematuria have a benign clinical course and do not require any specific therapy [5]. Once diagnosed, management involves regular clinical monitoring for the onset of hypertension, including routine blood pressure assessments. Laboratory follow-up should include serum creatinine, blood urea nitrogen levels, and urinalyses with urine protein-to-creatinine ratio measurements to monitor renal impairment and proteinuria progression. These routine evaluations can be performed by the primary care provider, with specialist referrals as needed every 1 to 2 years [2].
For our patients’ follow-up, urinalysis was conducted whenever urine discoloration was observed, especially after URI. Additionally, serial ultrasounds were performed to assess kidney size and any abnormal findings. If proteinuria or hypertension develops, in addition to lifestyle modifications, pharmacological therapy, involving the use of the maximum tolerated doses of renin-angiotensin-system inhibitors, such as angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, is recommended [6]. In this case, enalapril was prescribed as an angiotensin-converting enzyme inhibitor due to its superior potency, longer duration of action, and potentially improved safety profile compared to alternatives such as captopril [14].
Conclusion
This case highlights the importance of considering TBMD in pediatric patients presenting with recurrent macroscopic hematuria, particularly following URI. Although TBMD is often benign, it may present with atypical features and can be associated with proteinuria, necessitating careful evaluation. Differentiation from HSP, IgA nephropathy, and other glomerular diseases is crucial to ensure appropriate management. Given its hereditary nature, a comprehensive family history and genetic assessment should be considered. Long-term follow-up is essential for monitoring renal function, proteinuria progression, and managing hypertension, thereby preventing potential complications.
Study limitations
In this study, we did not encounter significant limitations. The only issue was the necessity of obtaining family consent for the kidney biopsy of the child, which, due to sensitivities and the family’s reluctance, was delayed for approximately 20 months, disrupting the patient’s diagnosis and treatment course.
Ethical Considerations
Compliance with ethical guidelines
All procedures performed in studies involving human participants followed the ethical standards of the Ethics Committee of Shahid Beheshti University of Medical Sciences, Tehran, Iran. Informed consent was obtained from the patient described in this case report.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualisation and study design: Masoumeh Mohkam and Neda Ghorbani-Khosroshahi; Data acquisition: Neda Ghorbani-Khosroshahi; Writing the original draft: Ghazal Azad and Neda Ghorbani-Khosroshahi, and Fatemeh Nili; Review and editing: Ghazal Azad, Neda Ghorbani-khosroshahi, and Masoumeh Mohkam; Supervision: Masoumeh Mohkam.
Conflicts of interest
The authors declared no conflict of interest.
Acknowledgements
The authors thank all patients who participated in the study. The authors also would like to express their gratitude to the staff of the Pediatric Nephrology Research Center of Mofid Children's Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
References
- Gattineni J. Highlights for the management of a child with proteinuria and hematuria. Int J Pediatr. 2012; 2012:768142. [DOI:10.1155/2012/768142] [PMID]
- Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003; 64(4):1169-78. [DOI:10.1046/j.1523-1755.2003.00234.x] [PMID]
- Pečovnik A, Haler ŽV, Wechtersbach K, Pleško J, Frelih M, Lindič J, et al. Histomorphological and clinical characteristics of patients with thin glomerular basement membrane nephropathy. Clin Nephrol. 2021; 96(1):24-30.[DOI:10.5414/CNP96S04] [PMID]
- Williams CEC, Lamond M, Marro J, Chetwynd AJ, Oni L. A narrative review of potential drug treatments for nephritis in children with IgA vasculitis (HSP). Clin Rheumatol. 2023; 42(12):3189-200. [DOI:10.1007/s10067-023-06781-8] [PMID]
- Uzzo M, Moroni G, Ponticelli C. Thin basement membrane: An underrated cause of end-stage renal disease. Nephron. 2023; 147(7):383-91. [DOI:10.1159/000528243] [PMID]
- Tryggvason K, Patrakka J. Thin basement membrane nephropathy. J Am Soc Nephrol. 2006; 17(3):813-22. [DOI:10.1681/ASN.2005070737] [PMID]
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int. 2001; 59(6):2069-72. [DOI:10.1046/j.1523-1755.2001.00721.x] [PMID]
- Yuste C, Gutierrez E, Sevillano AM, Rubio-Navarro A, Amaro-Villalobos JM, Ortiz A, et al. Pathogenesis of glomerular haematuria. World J Nephrol. 2015; 4(2):185-95.[DOI:10.5527/wjn.v4.i2.185] [PMID]
- Imam AA, Saadeh SA. Evaluation of Proteinuria and Hematuria in Ambulatory Setting. Pediatr Clin North Am. 2022; 69(6):1037-49. [DOI:10.1016/j.pcl.2022.07.002] [PMID]
- Nieuwhof CM, de Heer F, de Leeuw P, van Breda Vriesman PJ. Thin GBM nephropathy: Premature glomerular obsolescence is associated with hypertension and late onset renal failure. Kidney Int. 1997; 51(5):1596-601. [DOI:10.1038/ki.1997.219] [PMID]
- Piqueras AI, White RH, Raafat F, Moghal N, Milford DV. Renal biopsy diagnosis in children presenting with haematuria. Pediatr Nephrol. 1998; 12(5):386-91. [DOI:10.1007/s004670050471] [PMID]
- Nogueira M, Cartwright J Jr, Horn K, Doe N, Shappell S, Barrios R, et al. Thin basement membrane disease with heavy proteinuria or nephrotic syndrome at presentation. Am J Kidney Dis. 2000; 35(4):E15. [DOI:10.1016/S0272-6386(00)70033-3] [PMID]
- Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013; 24(3):364-75. [DOI:10.1681/ASN.2012020148] [PMID]
- Rumboldt Z, Marinković M, Drinovec J. Enalapril versus captopril: A double-blind multicentre comparison in essential hypertension. Int J Clin Pharmacol Res. 1988; 8(3):181-8. [PMID]
Type of Study: Case & Review |
Subject:
Pediatric Nephrology
Received: 2025/01/23 | Accepted: 2025/03/15 | Published: 2025/04/1
Received: 2025/01/23 | Accepted: 2025/03/15 | Published: 2025/04/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
