
Sat, Jul 5, 2025


Volume 7, Issue 2 (4-2019)
J. Pediatr. Rev 2019, 7(2): 113-120 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Vakili R, Hashemian S. Primordial Dwarfism: A Case Series From North East of Iran and Literature Review. J. Pediatr. Rev 2019; 7 (2) :113-120
URL: http://jpr.mazums.ac.ir/article-1-183-en.html
URL: http://jpr.mazums.ac.ir/article-1-183-en.html



1- Department of Pediatrics, Faculty of Medicine, Imam Reza Hospital, Mashhad University of Medical Sciences, Mashhad, Iran.
2- Department of Pediatrics, Faculty of Medicine, Imam Reza Hospital, Mashhad University of Medical Sciences, Mashhad, Iran. ,hashemians951@mums.ac.ir
2- Department of Pediatrics, Faculty of Medicine, Imam Reza Hospital, Mashhad University of Medical Sciences, Mashhad, Iran. ,
Full-Text [PDF 876 kb]
(4870 Downloads)
| Abstract (HTML) (8161 Views)

4. Review of Literature
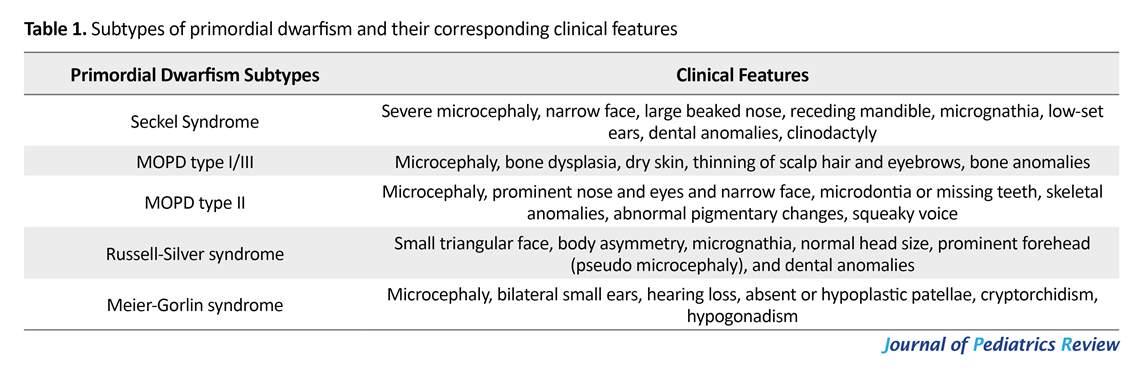
We presented seven PD cases who were referred to our center with prenatal and postnatal growth delay, without any specific diseases. PD is an incurable group of disorders caused by various mechanisms, presenting with growth deficiency, facial dysmorphism, and brain anomalies, and results in very small body size, over life (8). Growth delay continues after birth and causes short stature. In addition, PD is a highly heterogeneous condition with different clinical features that can be classified into 5 major subtypes including Seckel syndrome, RSS, microcephalic osteodysplastic primordial dwarfism types I/III and II Meier-Gorlin syndrome (1).
Seckel Syndrome (SS) is characterized by severe microcephaly, large beaked nose, mild to moderate mental retardation and called it bird-headed dwarfism (5). Narrow face, dental anomalies (9-10), receding mandible, micrognathia, scoliosis, hip dislocation, delayed bone age, clinodactyly, low-set ear, sternal abnormalities and seizure are other signs and symptoms (1, 11). Dislocation of eye lens, hypopigmented macules and Morgagni hernia are reported in rare cases (1). MODP types I, III are rare disorders with autosomal recessive inheritance (12). The characteristic features are growth retardation, bone dysplasia, and central nervous anomalies (13), dry skin, thinning of scalp hair and eyebrows (1).
Patients with MODP I may have microcephaly, apnea, seizure, corpus callosum agenesis, short vertebra, bent femur and hip displacement. The signs of other subtypes of MOPD III are intrauterine growth retardation, clavicles and bone anomalies (1). Investigations found evidence that MOPD I and III are variations of one type and should be classified together, despite their radiologic and physical differences (13). Majewski Osteodysplastic Primordial Dwarfism type II (MOPD) is a distinct disorder. This rare autosomal recessive condition is estimated to be the most common type of PD (14).
Patients are characterize by IUGR, very low birth weight (less than 1500 g) high-pitched voice (squeaky voice), prominent nose and eyes and narrow face, microdontia or missing teeth, skeletal anomalies such as delayed bone age, thin bones, coxavara, small iliac wings, flat acetabular angle, hip dislocation at birth, short first metacarpals, carpal bones fusion (6), borderline intellectual functioning or mild mental retardation, abnormal pigmentary changes (1, 14), problems in 80% of infants, and truncal obesity that may accelerate the puberty (15).
Another important feature of MOPD II is the early onset of cerebrovascular diseases such as cerebral aneurysm and occolusive arteriopathy (moyamoya disease) (16). Hematologic abnormalities such as anemia, leukocytosis and thrombocytosis may occur in affected patients (1, 15). The characteristic features of Russell-Silver syndrome are low birth weight, short stature, body asymmetry (17, 18), and normal head size (1). Other diagnostic features are small triangular face, micrognathia, dental anomalies, prominent forehead that causes pseudo microcephaly (19). Feeding difficulties such as malnutrition, gastrointestinal reflux and vomiting are frequent signs in infants and children with RSS (20).
Meier-Gorlin syndrome (ear-patella-short stature syndrome) is another type of PD that was first described in 1959, in which Meier et al. described a case with micrognathia, microtia, absent patellae, and cryptorchidism (7). The major diagnostic criteria of this syndrome include bilateral small ears, aplasia or hypoplasia of patellae, and short stature with normal mentality (21). Other clinical features include microcephaly, lower limb arthrogryposis, cryptorchidism, hypogonadism, deafness, curved clavicle and deformed ribs (1, 19).
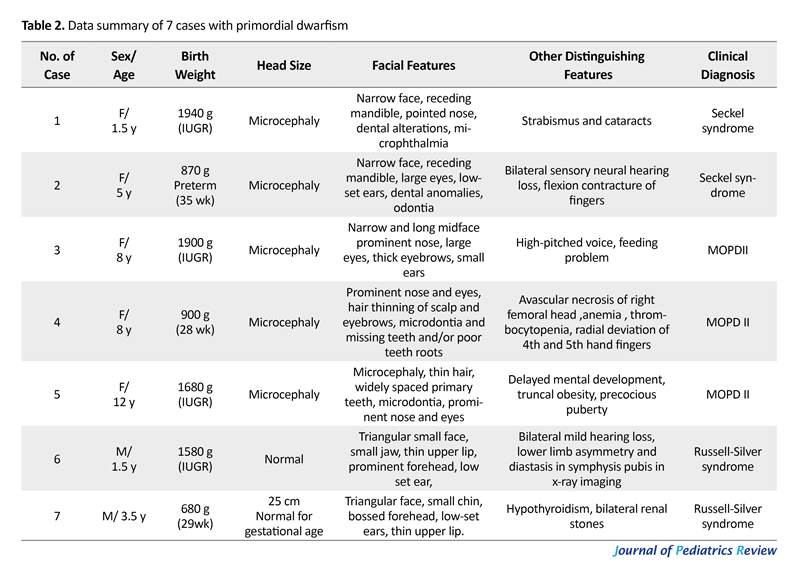
We reported 7 patients with severe progressing growth deficiency from birth. The diagnosis was made based on severe growth retardation, specific clinical findings and ruling out other similar situations. Laboratory tests were assessed in all cases to differentiate apparently similar disorders with primordial dwarfism, such as Fanconi anemia, Bloom syndrome, 3M syndrome, etc. (1, 18). In laboratory examination, one of the MOPD II cases showed anemia and thrombocytosis after 2 years fallow up (case 4). However, other tests were normal in all patients. Chromosomal studies were normal in all cases.
Radiologic studies showed delayed bone age in all patients. Other findings included thin bones and Avascular Necrosis (AVN) of right head of femur (AVN type 3) in our case 4. AVN and hip instability may occur in underlying disorders or growth hormone therapy in PD patients. Another finding was carpal bones fusion, shown in Figure 2. Brain imaging showed no evidence for cerebrovascular diseases. Cerebrovascular diseases occur in 32% of MOPD II patients. Occlusive arteriopathy can be clinically silent and occur in younger individuals (16). Different clinical findings in PD patients can be due to mutations of various genes (3). Also, chromosomal studies have reported specific changes in different subtypes of PD (1). Findings of molecular, genetic and chromosomal changes can be useful to differentiate PD subtypes from each other.
This may help in screening and prenatal diagnosis of families in future. Management of PD patients is a problem when the objective is improvement of patient’s final stature. There are no effective treatment available for some aspects of primordial dwarfism. Growth studies revealed that human Gowth Hormone (GH) therapy can improve final height and result in a positive growth response (22). Many investigations estimated the effect of GH therapy on different types of PD. Most of these studies reported no improvement in final stature, following GH therapy in MOPD type II patients. There is no study on MOPD types I/III (1). Birbaek et al. demonstrated improvement in growth rate after GH therapy in Seckel cases (1); however, it is not approved by other studies. Also, Munnik et al. reported an increase in growth rate following GH therapy in Meire–Gorlin syndrome (1).
RSS is an indication for GH therapy under the SGA registered license in the USA and European Medicine Agency. Clinical trials approved GH therapy for patients with RSS (23). We have applied GH treatment in 2 of our cases with RSS since 4 months ago. Both of them responded to GH therapy with an increase of 3-4 cm in height (length for head z score improved from -5.7 to -4.8 in case 6 and from -5 to -4.2 in case of 7), that showed benefit in our patients. Studies revealed that GH treatment can be effective for some subtypes of PD. However further studies are required to appoint the indications of GH therapy for primordial dwarfism patients.
This review and case presentation study aimed to present an overview of clinical features of PD patients. However, we could not confirm these findings with genetic analysis. Therefore, further studies are required to reveal molecular and chromosomal alterations, in order to discuss overall characteristics of this disorder.
Ethical Considerations
Compliance with ethical guidelines
There is no ethical principle to be considered doing this research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors contributions
All authors have read and approved the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
Full-Text: (6354 Views)
1. Introduction
rimordial Dwarfism (PD) is a group of disorders with significant prenatal and postnatal growth retardation and various clinical abnormalities that differ from one type to another (1). PD is characterized by intrauterine growth retardation, low birth weight, and Small (bones and body organs) for Gestational Age (SGA). Female and male patients are equally affected and look smaller in size (weight and height) compared to normal individuals (2). The main specific feature of PD that differentiates it from other forms of dwarfism is reduced head size in proportion to body, called “microcephalic primordial dwarfism.” There is only 1 subtype of PD with normal head size, called Russell-Silver Syndrome (RSS). Clinical findings vary from one subtype to another. Molecular, genetic and chromosomal changes can contribute in development of PD (3).
PD is a rare heterogeneous condition and diagnostic category including specific features in 5 major subtypes. The main feature in all subtypes is prenatal and postnatal growth retardation. Patients are very small for their age and sex. They are diagnosed with Intrauterine Growth Retardation (IUGR) and often have premature delivery and low birth weight (SGA). Growth delay continues after birth (1).
Five subtypes in PD include Seckel syndrome, Majewski Osteodysplastic Primordial Dwarfism (MOPD type I/III), MOPD type II, Meier-Gorlin syndrome and Russell-Silver Syndrome (1). Russell-Silver syndrome is a specific group of patients with normal head size; other types have microcephaly (3). Seckel Syndrome (SS) is a rare autosomal recessive disorder (4) and first defined by Seckel in 1960. He described 2 case studies by himself and 13 cases documented in the literature, over a 200-year period, characterized by severe microcephaly, large beaked nose, and mild to moderate mental retardation and called it bird-headed dwarfism (5).
In 1982, Majewski et al. introduced the term osteodysplastic primordial dwarfism through literature review. They classified it in 3 categories according to growth and mental retardation, bony anomalies and specific radiologic findings (6). Russell-Silver syndrome was first reported by Russell and Silver in the 1950’s and 1960’s. The characteristic features of Russell-Silver syndrome are low birth weight, short stature, body asymmetry and normal head size (1). Meier-Gorlin syndrome (ear-patella-short stature syndrome) is another type of PD that is an autosomal recessive disorder. Meier et al. first described a case with micrognathia, microtia, absent patellae and cryptorchidism in 1959 (7). Further clinical diagnostic criteria of these subtypes are presented in Table 1.
We presented 7 patients with different clinical presentations of this disorder. All cases had growth retardation from birth and other clinical features that helped us to diagnose the disorder described below. The current study aimed to present this disorder in Iran with its different types for the first time. Genetic samples were not included in this study due to limitations in genetic assays and the expenses of that, in our country. We present these cases of primordial dwarfism and their clinical features as the first time for clinicians and other health care groups.
2. Case Presentation
2.1. Case 1
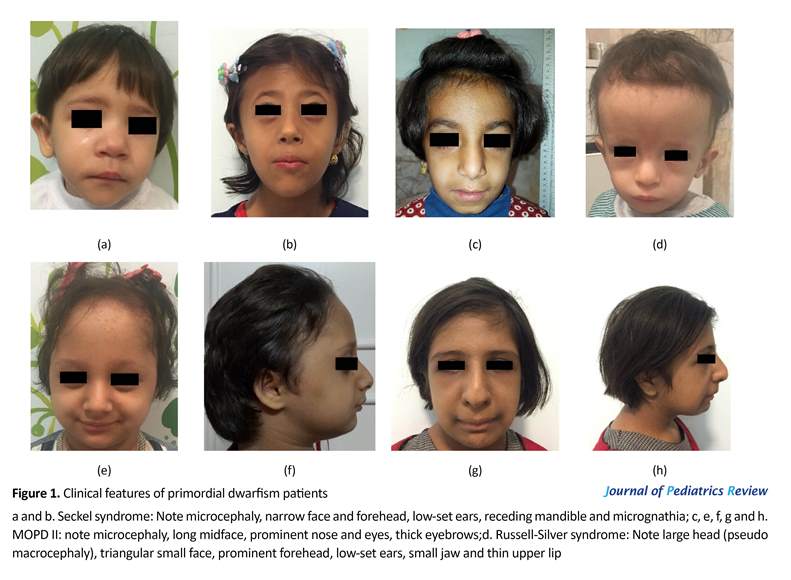
An 18-month-old girl was the second born child of non-consanguineous, healthy parents. She was born at 40 weeks of gestation. Pregnancy was complicated by Intrauterine Growth Retardation (IUGR). Birth weight was 1940 g, height was 42 cm and head circumference was 31 cm, all below the third percentile. Physical examination revealed dysmorphic features, microcephaly, narrow face, receding mandible, pointed nose, dental alterations, microphthalmia (Figure 1a). Other findings were developmental delay in speech and motor skills, as well as strabismus and cataract in ophthalmologic tests. Her older sister had normal physical appearance and mental growth. A diagnosis of Seckel syndrome was made based on these clinical findings.
2.2. Case 2
A 5-year-old girl who was the first born child of non-consanguineous parents who was born at preterm with gestational age of 35 weeks and IUGR delivery. Her birth weight was 870 g, her head circumference was 25 cm and birth height was 44 cm, all under the third percentile. Her latest weight was 10 kg (-6.2 SDS [Standard Deviation Score]), with head circumference of 36 cm and height of 92 cm (-3.6 SDS). According to the clinical findings, she had severe growth retardation, microcephaly, narrow face, receding mandible, large eyes, dental anomalies and odonthia, bilateral sensory neural hearing loss, flexion contracture of hand finger and mild mental retardation. She was diagnosed with Seckel syndrome (Figure 1b).
rimordial Dwarfism (PD) is a group of disorders with significant prenatal and postnatal growth retardation and various clinical abnormalities that differ from one type to another (1). PD is characterized by intrauterine growth retardation, low birth weight, and Small (bones and body organs) for Gestational Age (SGA). Female and male patients are equally affected and look smaller in size (weight and height) compared to normal individuals (2). The main specific feature of PD that differentiates it from other forms of dwarfism is reduced head size in proportion to body, called “microcephalic primordial dwarfism.” There is only 1 subtype of PD with normal head size, called Russell-Silver Syndrome (RSS). Clinical findings vary from one subtype to another. Molecular, genetic and chromosomal changes can contribute in development of PD (3).
PD is a rare heterogeneous condition and diagnostic category including specific features in 5 major subtypes. The main feature in all subtypes is prenatal and postnatal growth retardation. Patients are very small for their age and sex. They are diagnosed with Intrauterine Growth Retardation (IUGR) and often have premature delivery and low birth weight (SGA). Growth delay continues after birth (1).
Five subtypes in PD include Seckel syndrome, Majewski Osteodysplastic Primordial Dwarfism (MOPD type I/III), MOPD type II, Meier-Gorlin syndrome and Russell-Silver Syndrome (1). Russell-Silver syndrome is a specific group of patients with normal head size; other types have microcephaly (3). Seckel Syndrome (SS) is a rare autosomal recessive disorder (4) and first defined by Seckel in 1960. He described 2 case studies by himself and 13 cases documented in the literature, over a 200-year period, characterized by severe microcephaly, large beaked nose, and mild to moderate mental retardation and called it bird-headed dwarfism (5).
In 1982, Majewski et al. introduced the term osteodysplastic primordial dwarfism through literature review. They classified it in 3 categories according to growth and mental retardation, bony anomalies and specific radiologic findings (6). Russell-Silver syndrome was first reported by Russell and Silver in the 1950’s and 1960’s. The characteristic features of Russell-Silver syndrome are low birth weight, short stature, body asymmetry and normal head size (1). Meier-Gorlin syndrome (ear-patella-short stature syndrome) is another type of PD that is an autosomal recessive disorder. Meier et al. first described a case with micrognathia, microtia, absent patellae and cryptorchidism in 1959 (7). Further clinical diagnostic criteria of these subtypes are presented in Table 1.
We presented 7 patients with different clinical presentations of this disorder. All cases had growth retardation from birth and other clinical features that helped us to diagnose the disorder described below. The current study aimed to present this disorder in Iran with its different types for the first time. Genetic samples were not included in this study due to limitations in genetic assays and the expenses of that, in our country. We present these cases of primordial dwarfism and their clinical features as the first time for clinicians and other health care groups.
2. Case Presentation
2.1. Case 1
An 18-month-old girl was the second born child of non-consanguineous, healthy parents. She was born at 40 weeks of gestation. Pregnancy was complicated by Intrauterine Growth Retardation (IUGR). Birth weight was 1940 g, height was 42 cm and head circumference was 31 cm, all below the third percentile. Physical examination revealed dysmorphic features, microcephaly, narrow face, receding mandible, pointed nose, dental alterations, microphthalmia (Figure 1a). Other findings were developmental delay in speech and motor skills, as well as strabismus and cataract in ophthalmologic tests. Her older sister had normal physical appearance and mental growth. A diagnosis of Seckel syndrome was made based on these clinical findings.
2.2. Case 2
A 5-year-old girl who was the first born child of non-consanguineous parents who was born at preterm with gestational age of 35 weeks and IUGR delivery. Her birth weight was 870 g, her head circumference was 25 cm and birth height was 44 cm, all under the third percentile. Her latest weight was 10 kg (-6.2 SDS [Standard Deviation Score]), with head circumference of 36 cm and height of 92 cm (-3.6 SDS). According to the clinical findings, she had severe growth retardation, microcephaly, narrow face, receding mandible, large eyes, dental anomalies and odonthia, bilateral sensory neural hearing loss, flexion contracture of hand finger and mild mental retardation. She was diagnosed with Seckel syndrome (Figure 1b).
2.3. Case 3
An 8-year-old girl, the first born child of consanguineous parents (first cousin parents) who had IUGR at birth (birth weight 1900 g), and microcephaly. Growth delay continued after birth. Physical examination revealed a narrow and long midface, high-pitched voice, prominent nose, large eyes, thick eyebrows and small ears. There was feeding problem in infancy. She had the signs of premature thelarche and pubarche at the age of 7 years and had mild mental retardation. She could not walk until the age of 2.5 years. Skeletal imaging showed permanent fusion of 2 wrist bones (Figure 1c and Figure 2a). She was diagnosed with MOPD II syndrome based on the clinical findings.
2.4. Case 4
An 8-year-old girl was referred to our department with severe growth retardation and microcephaly. She weighed 13 kg (-5.99 SDS). Her height was 92 cm (-7.2 SDS) and her head circumference was 43 cm. Birth history revealed that she was born at preterm (28 weeks). She was the second child of healthy parents (first cousin parents). Her birth weight was 900 g, her length was 30 cm, and head circumference was 20 cm. Pregnancy was complicated with bleeding, fetal decelerations, and premature delivery. A clinical examination revealed diagnostic features of PD in MOPD subgroup such as microcephaly, prominent nose and eyes, hair thinning of scalp hair and eyebrows, microdontia and missing teeth or poor tooth roots.
She was suffering from abnormality in ophthalmologic examination, high-pitched nasal voice, mild mental retardation and poor sleep patterns. She had some skeletal abnormalities such as radial deviation of the fourth and fifth hand fingers, and abnormal development of the hip and dysplasia. She had limping that made her undergo surgery and X-rays revealed avascular necrosis of right femoral head (Figure 1e, Figure 1f and Figure 2b). Puberty signs occurred early at the age of 7. Lab tests detected recently developed hematologic abnormalities in her (anemia and thrombocytosis). These findings helped us to diagnose MOPD II based on clinical features of such patients in other reports.
2.5. Case 5
A 12-year-old girl, second born child of consanguineous healthy parents (first cousin parents). Her height was 110 cm (-5.4 SDS), her weight was 16 kg (-7 SDS) and her occipital frontal circumferences was 44 cm. she had IUGR at birth with birth weight of 1680 g, birth length of 42 cm and birth head circumference of 27 cm. She exhibited microcephaly, thin hair, widely spaced primary teeth, microdontia, prominent nose and eyes, ophthalmologic disorders, delayed mental development, truncal obesity and precocious puberty (Figure 1g and Figure 1h). On the basis of these findings, she was diagnosed with MOPD II.
2.6. Case 6
An 18-month-old boy, the second born child of non-relative healthy parents, was IUGR at birth with birth weight of 1580 g, height of 41 cm, and head circumference of 35 cm. His growth delay continued after birth; however, head circumference was below the normal range. His latest weight was 5500 g (-8 SDS), his height was 68 cm (-3.9 SDS), and his circumference was 46 cm. Clinical findings revealed a large head compared to rest of the body (pseudo macrocephaly), triangular small face, small jaw, thin upper lip, prominent forehead, low-set ears, bilateral mild hearing loss. X-ray imaging detected lower limb asymmetry and diastasis symphysis pubis (Figure 1d and Figure 2c). She also suffered from lack of appetite, developmental delay in motor skills and delayed bone age (9 months). Furthermore, he had inguinal hernia in infancy. These clinical features and normal head size can be signs of Russell-Silver syndrome.
2.7. Case 7
A 3.5-year-old boy, the first child of healthy parents, was born at 29 weeks of pregnancy. His birth weight was 680 g, height was 33 cm and head circumference was 25 cm. Growth was delayed after birth, except for head size. His weight was 10 kg (-4.4 SDS) and height 80 cm (-5.SDS) in 3.5 years old. Other clinical features included triangular face, small chin, bossed forehead, low-set ears and thin upper lip. He suffered from hypothyroidism and bilateral renal stones that was under medication. These clinical appearances helped us to diagnose him with Russell-Silver syndrome.
We reviewed 7 cases with clinical features of growth insufficiency including prenatal and postnatal growth delay and suspicious signs of primordial dwarfism. The current investigation was the first case series of PD cases in Iran. Therefore, we conducted a literature review on primordial dwarfism through Google Scholar, Medline, and PubMed data bases, to compare our results with previously reported cases. PD is a rare disorder with a spectrum of clinical conditions. Therefore, we selected articles that could help us explain the clinical symptoms and sings of PD.
3. Discussion
We reported 7 patients with prenatal and postnatal growth retardation and diagnostic clinical features of primordial dwarfism. Our cases were either small for their gestational age at birth, or premature. Their postnatal growth rate was very slow. Our diagnosis of PD was based on severe growth retardation at birth and afterwards and special clinical features that helped us classify them into subgroups of PD. Different laboratory tests have been evaluated in all cases to differentiate overlapping disorders with primordial dwarfism, such as Fanconi anemia, Bloom syndrome, 3M syndrome, and so on (1).
Laboratory tests included complete blood count, liver and renal function tests, TSH and T4, serum insulin and glucose, LH and FSH, growth hormone levels, and immunologic tests. The wrist X-rays were taken for estimating bone age in all patients. Additional evaluation were accomplished according to the primary diagnosis of other centers, including abnormalities in abdominal ultrasonography and brain MRI of the patients.
The cases are presented on the basis of specific clinical appearance of PD. Reports from different countries present these cases by different methods. Some studies applied genetic and molecular analysis to confirm them. However, we could not use genetic assays, because they are not available everywhere in Iran. In detail, we presented 2 cases of Seckel syndrome, 3 cases of MOPD II and 2 cases of RSS. Parents agreed to present their children in our study. We studied 2 other cases whom their parents did not agree to report their children, thus we excluded them from the case presentation. Data summary of these 7 patients are listed in Table 2.
An 8-year-old girl, the first born child of consanguineous parents (first cousin parents) who had IUGR at birth (birth weight 1900 g), and microcephaly. Growth delay continued after birth. Physical examination revealed a narrow and long midface, high-pitched voice, prominent nose, large eyes, thick eyebrows and small ears. There was feeding problem in infancy. She had the signs of premature thelarche and pubarche at the age of 7 years and had mild mental retardation. She could not walk until the age of 2.5 years. Skeletal imaging showed permanent fusion of 2 wrist bones (Figure 1c and Figure 2a). She was diagnosed with MOPD II syndrome based on the clinical findings.
2.4. Case 4
An 8-year-old girl was referred to our department with severe growth retardation and microcephaly. She weighed 13 kg (-5.99 SDS). Her height was 92 cm (-7.2 SDS) and her head circumference was 43 cm. Birth history revealed that she was born at preterm (28 weeks). She was the second child of healthy parents (first cousin parents). Her birth weight was 900 g, her length was 30 cm, and head circumference was 20 cm. Pregnancy was complicated with bleeding, fetal decelerations, and premature delivery. A clinical examination revealed diagnostic features of PD in MOPD subgroup such as microcephaly, prominent nose and eyes, hair thinning of scalp hair and eyebrows, microdontia and missing teeth or poor tooth roots.
She was suffering from abnormality in ophthalmologic examination, high-pitched nasal voice, mild mental retardation and poor sleep patterns. She had some skeletal abnormalities such as radial deviation of the fourth and fifth hand fingers, and abnormal development of the hip and dysplasia. She had limping that made her undergo surgery and X-rays revealed avascular necrosis of right femoral head (Figure 1e, Figure 1f and Figure 2b). Puberty signs occurred early at the age of 7. Lab tests detected recently developed hematologic abnormalities in her (anemia and thrombocytosis). These findings helped us to diagnose MOPD II based on clinical features of such patients in other reports.
2.5. Case 5
A 12-year-old girl, second born child of consanguineous healthy parents (first cousin parents). Her height was 110 cm (-5.4 SDS), her weight was 16 kg (-7 SDS) and her occipital frontal circumferences was 44 cm. she had IUGR at birth with birth weight of 1680 g, birth length of 42 cm and birth head circumference of 27 cm. She exhibited microcephaly, thin hair, widely spaced primary teeth, microdontia, prominent nose and eyes, ophthalmologic disorders, delayed mental development, truncal obesity and precocious puberty (Figure 1g and Figure 1h). On the basis of these findings, she was diagnosed with MOPD II.
2.6. Case 6
An 18-month-old boy, the second born child of non-relative healthy parents, was IUGR at birth with birth weight of 1580 g, height of 41 cm, and head circumference of 35 cm. His growth delay continued after birth; however, head circumference was below the normal range. His latest weight was 5500 g (-8 SDS), his height was 68 cm (-3.9 SDS), and his circumference was 46 cm. Clinical findings revealed a large head compared to rest of the body (pseudo macrocephaly), triangular small face, small jaw, thin upper lip, prominent forehead, low-set ears, bilateral mild hearing loss. X-ray imaging detected lower limb asymmetry and diastasis symphysis pubis (Figure 1d and Figure 2c). She also suffered from lack of appetite, developmental delay in motor skills and delayed bone age (9 months). Furthermore, he had inguinal hernia in infancy. These clinical features and normal head size can be signs of Russell-Silver syndrome.
2.7. Case 7
A 3.5-year-old boy, the first child of healthy parents, was born at 29 weeks of pregnancy. His birth weight was 680 g, height was 33 cm and head circumference was 25 cm. Growth was delayed after birth, except for head size. His weight was 10 kg (-4.4 SDS) and height 80 cm (-5.SDS) in 3.5 years old. Other clinical features included triangular face, small chin, bossed forehead, low-set ears and thin upper lip. He suffered from hypothyroidism and bilateral renal stones that was under medication. These clinical appearances helped us to diagnose him with Russell-Silver syndrome.
We reviewed 7 cases with clinical features of growth insufficiency including prenatal and postnatal growth delay and suspicious signs of primordial dwarfism. The current investigation was the first case series of PD cases in Iran. Therefore, we conducted a literature review on primordial dwarfism through Google Scholar, Medline, and PubMed data bases, to compare our results with previously reported cases. PD is a rare disorder with a spectrum of clinical conditions. Therefore, we selected articles that could help us explain the clinical symptoms and sings of PD.
3. Discussion
We reported 7 patients with prenatal and postnatal growth retardation and diagnostic clinical features of primordial dwarfism. Our cases were either small for their gestational age at birth, or premature. Their postnatal growth rate was very slow. Our diagnosis of PD was based on severe growth retardation at birth and afterwards and special clinical features that helped us classify them into subgroups of PD. Different laboratory tests have been evaluated in all cases to differentiate overlapping disorders with primordial dwarfism, such as Fanconi anemia, Bloom syndrome, 3M syndrome, and so on (1).
Laboratory tests included complete blood count, liver and renal function tests, TSH and T4, serum insulin and glucose, LH and FSH, growth hormone levels, and immunologic tests. The wrist X-rays were taken for estimating bone age in all patients. Additional evaluation were accomplished according to the primary diagnosis of other centers, including abnormalities in abdominal ultrasonography and brain MRI of the patients.
The cases are presented on the basis of specific clinical appearance of PD. Reports from different countries present these cases by different methods. Some studies applied genetic and molecular analysis to confirm them. However, we could not use genetic assays, because they are not available everywhere in Iran. In detail, we presented 2 cases of Seckel syndrome, 3 cases of MOPD II and 2 cases of RSS. Parents agreed to present their children in our study. We studied 2 other cases whom their parents did not agree to report their children, thus we excluded them from the case presentation. Data summary of these 7 patients are listed in Table 2.
4. Review of Literature
We presented seven PD cases who were referred to our center with prenatal and postnatal growth delay, without any specific diseases. PD is an incurable group of disorders caused by various mechanisms, presenting with growth deficiency, facial dysmorphism, and brain anomalies, and results in very small body size, over life (8). Growth delay continues after birth and causes short stature. In addition, PD is a highly heterogeneous condition with different clinical features that can be classified into 5 major subtypes including Seckel syndrome, RSS, microcephalic osteodysplastic primordial dwarfism types I/III and II Meier-Gorlin syndrome (1).
Seckel Syndrome (SS) is characterized by severe microcephaly, large beaked nose, mild to moderate mental retardation and called it bird-headed dwarfism (5). Narrow face, dental anomalies (9-10), receding mandible, micrognathia, scoliosis, hip dislocation, delayed bone age, clinodactyly, low-set ear, sternal abnormalities and seizure are other signs and symptoms (1, 11). Dislocation of eye lens, hypopigmented macules and Morgagni hernia are reported in rare cases (1). MODP types I, III are rare disorders with autosomal recessive inheritance (12). The characteristic features are growth retardation, bone dysplasia, and central nervous anomalies (13), dry skin, thinning of scalp hair and eyebrows (1).
Patients with MODP I may have microcephaly, apnea, seizure, corpus callosum agenesis, short vertebra, bent femur and hip displacement. The signs of other subtypes of MOPD III are intrauterine growth retardation, clavicles and bone anomalies (1). Investigations found evidence that MOPD I and III are variations of one type and should be classified together, despite their radiologic and physical differences (13). Majewski Osteodysplastic Primordial Dwarfism type II (MOPD) is a distinct disorder. This rare autosomal recessive condition is estimated to be the most common type of PD (14).
Patients are characterize by IUGR, very low birth weight (less than 1500 g) high-pitched voice (squeaky voice), prominent nose and eyes and narrow face, microdontia or missing teeth, skeletal anomalies such as delayed bone age, thin bones, coxavara, small iliac wings, flat acetabular angle, hip dislocation at birth, short first metacarpals, carpal bones fusion (6), borderline intellectual functioning or mild mental retardation, abnormal pigmentary changes (1, 14), problems in 80% of infants, and truncal obesity that may accelerate the puberty (15).
Another important feature of MOPD II is the early onset of cerebrovascular diseases such as cerebral aneurysm and occolusive arteriopathy (moyamoya disease) (16). Hematologic abnormalities such as anemia, leukocytosis and thrombocytosis may occur in affected patients (1, 15). The characteristic features of Russell-Silver syndrome are low birth weight, short stature, body asymmetry (17, 18), and normal head size (1). Other diagnostic features are small triangular face, micrognathia, dental anomalies, prominent forehead that causes pseudo microcephaly (19). Feeding difficulties such as malnutrition, gastrointestinal reflux and vomiting are frequent signs in infants and children with RSS (20).
Meier-Gorlin syndrome (ear-patella-short stature syndrome) is another type of PD that was first described in 1959, in which Meier et al. described a case with micrognathia, microtia, absent patellae, and cryptorchidism (7). The major diagnostic criteria of this syndrome include bilateral small ears, aplasia or hypoplasia of patellae, and short stature with normal mentality (21). Other clinical features include microcephaly, lower limb arthrogryposis, cryptorchidism, hypogonadism, deafness, curved clavicle and deformed ribs (1, 19).
We reported 7 patients with severe progressing growth deficiency from birth. The diagnosis was made based on severe growth retardation, specific clinical findings and ruling out other similar situations. Laboratory tests were assessed in all cases to differentiate apparently similar disorders with primordial dwarfism, such as Fanconi anemia, Bloom syndrome, 3M syndrome, etc. (1, 18). In laboratory examination, one of the MOPD II cases showed anemia and thrombocytosis after 2 years fallow up (case 4). However, other tests were normal in all patients. Chromosomal studies were normal in all cases.
Radiologic studies showed delayed bone age in all patients. Other findings included thin bones and Avascular Necrosis (AVN) of right head of femur (AVN type 3) in our case 4. AVN and hip instability may occur in underlying disorders or growth hormone therapy in PD patients. Another finding was carpal bones fusion, shown in Figure 2. Brain imaging showed no evidence for cerebrovascular diseases. Cerebrovascular diseases occur in 32% of MOPD II patients. Occlusive arteriopathy can be clinically silent and occur in younger individuals (16). Different clinical findings in PD patients can be due to mutations of various genes (3). Also, chromosomal studies have reported specific changes in different subtypes of PD (1). Findings of molecular, genetic and chromosomal changes can be useful to differentiate PD subtypes from each other.
This may help in screening and prenatal diagnosis of families in future. Management of PD patients is a problem when the objective is improvement of patient’s final stature. There are no effective treatment available for some aspects of primordial dwarfism. Growth studies revealed that human Gowth Hormone (GH) therapy can improve final height and result in a positive growth response (22). Many investigations estimated the effect of GH therapy on different types of PD. Most of these studies reported no improvement in final stature, following GH therapy in MOPD type II patients. There is no study on MOPD types I/III (1). Birbaek et al. demonstrated improvement in growth rate after GH therapy in Seckel cases (1); however, it is not approved by other studies. Also, Munnik et al. reported an increase in growth rate following GH therapy in Meire–Gorlin syndrome (1).
RSS is an indication for GH therapy under the SGA registered license in the USA and European Medicine Agency. Clinical trials approved GH therapy for patients with RSS (23). We have applied GH treatment in 2 of our cases with RSS since 4 months ago. Both of them responded to GH therapy with an increase of 3-4 cm in height (length for head z score improved from -5.7 to -4.8 in case 6 and from -5 to -4.2 in case of 7), that showed benefit in our patients. Studies revealed that GH treatment can be effective for some subtypes of PD. However further studies are required to appoint the indications of GH therapy for primordial dwarfism patients.
This review and case presentation study aimed to present an overview of clinical features of PD patients. However, we could not confirm these findings with genetic analysis. Therefore, further studies are required to reveal molecular and chromosomal alterations, in order to discuss overall characteristics of this disorder.
Ethical Considerations
Compliance with ethical guidelines
There is no ethical principle to be considered doing this research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors contributions
All authors have read and approved the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- Khaetarpal P, Das Kh, Panigrahi I, Munshi A. Primoral dwarfism: Overview of clinical and genetic aspects. Molecular Genetics and Genomics. 2016; 291(1):1-15. [DOI:10.1007/s00438-015-1110-y] [PMID])
- Codd PJ, Scott RM, Smith ER. Seckel syndrome and moyamoya: Case report. Journal of Neurosurgery: Pediatrics. 2009; 3(4):320-4. [DOI:10.3171/2008.12.PEDS08205] [PMID]
- Klingsein A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Development. 2011; 25(19):2011-24. [DOI:10.1101/gad.169037] [PMID] [PMCID]
- Filonava L, Torres AG, de Pouplana LR. A novel cause for primordial dwarfism revealed: Defective tRNA modification. Genome Biology. 2015; 16(1):216.
- Vardhan BH, Muthu MS, Saraswathi K, Koteeswaran D. Bird-headed dwarf of Seckel. Journal of Indian Society of Pedodontics and Preventive Dentistry. 2007; 25(5):8-9.
- Seckel HP, editor. Bird-headed dwarfs: Studies in developmental anthropology including human proportions. Pediatrics. 1961; 27(3):426-9.
- Seymen F, Tuna b, Kayserili H. Seckel syndrome: Report accuses. Journal of Clinical Pediatric Dentistry. 2002; 26(3):305-9.
- Kjær I, Hansen N, Becktor KB, Birkebaek N, Balslev T. Craniofacial morphology, dentition, and skeletal maturity in four siblings with seckel syndrome. The Cleft Palate-Craniofacial Journal. 2001; 38(6):645-51. [DOI:10.1597/1545-1569_2001_038_0645_cmdasm_2.0.co_2]
- Mokrani Benhelli H, Gaillard L, Biasutto P, Guen T, Touzot F, Vasquez N, et al. Primary microcephaly, impaired DNA replication and genomic instability caused by compound heterozygous ATR mutations. Human Mutation. 2013; 34(2):374-84. [DOI:10.1002/humu.22245] [PMID]
- Majweski F, Goecke T. Studies of microcephalic primordial dwarfism I: Approach to a delineation of the seckle syndrome. American Journal of Medical Genetics. 1982; 12(1):7-21. [DOI:10.1002/ajmg.1320120103] [PMID]
- Meinecke P, Passarge E. Microcephalic osteodysplastic primordial dwarfism type I/II in sibs. Journal of Medical Genetics. 1991; 28(11):795-800. [DOI:10.1136/jmg.28.11.795]
- Melinda J, Pierce RP, Morse RP. The neurologic findings in Taybi- Linder syndrome (MOPD I/II): Case report and review of the literature. American Journal of Medical genetics. 2012; 158(3):606-10. [DOI:10.1002/ajmg.a.33958] [PMID]
- Rauch A. The shortest of the short: Pericentrin mutations and beyond. Best Practice & Research Clinical Endocrinology & Metabolism. 2011; 25(1):125-30. [DOI:10.1016/j.beem.2010.10.015] [PMID]
- Hall JG, Flora C, Scott CI, Pauli RM, Tanaka KI. Majweski osteodysplastic primordial dwarfism type II (MOPD II): Natural history and clinical findings. American Journal of Medical Genetics. 2004; 130(1):55-72. [DOI:10.1002/ajmg.a.30203] [PMID]
- Luck D, Robertson F, Ganesan V. Screening for cerebrovascular disease in Microcephalic Osteodyspalstic Primordial Dwarfism type II (MOPD II): An evidence- based proposal. Pediatric Neurology. 2013; 48(4):294-8. [DOI:10.1016/j.pediatrneurol.2012.12.010] [PMID]
- Kent T, Yamaguchi Jr, Jennifer B, Salam MA, Karen S, Myung MD, et al. Spinal deformity in Russell-silver syndrome. Spinal Deformity. 2015; 3(1):95-7. [DOI:10.1016/j.jspd.2014.06.003] [PMID]
- Rao VB, Lily K, Seema K, Ghosh K, Dipika M. Paternal reciprocal translocation t(11;16)(p13;q24.3) in a Silver–Russel syndrome patient. Annales de Génétique. 2003; 46(4):475-8. [DOI:10.1016/S0003-3995(03)00028-5]
- Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu Amero SN, et al. Exploring the spectrum of 3‐M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clinical Endocrinology. 2012; 77(3):335-42. [DOI:10.1111/j.1365-2265.2012.04428.x]
- Marsaud C, Rossingnol S, Tounian P, Netchine I, Dubern B. Prevalence and management of gastro intestinal manifestation in silver- Russell syndrome. Archives of Disease in Childhood. 2015; 100(4):353-8. [DOI:10.1136/archdischild-2013-305864] [PMID]
- Sonja A, De Munnik, Hoesloot E, Roukema J, Schoots J, Kneors N, et al. Meier- Gorlin syndrome. Orphanet Journal of Rare Diseases. 2015; 10:114. [DOI:10.1186/s13023-015-0322-x] [PMID] [PMCID]
- Shawky RM, Gamal R. Meier–Gorlin syndrome: An additional Egyptian patient with gastroesophageal reflux, hydronephrosis, renal stones and hypoplastic labia majora and minora with clitromegaly. Egyptian Journal of Medical Human Genetics. 2016; 17(4):397-400.
- Ranke MB, Lindberg A, Mullis PE, Geffner ME, Tanaka T, Cutfield WS, et al. Towards optimal treatment with growth hormone in short children and adolescents: Evidence and theses. Hormone Research in Pædiatrics. 2013; 79(2):51-67. [DOI:10.1159/000347121] [PMID]
- Wakeling EL, Brioude F, Lokulo Sodipe O, O’connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nature Reviews Endocrinology. 2017; 13(2):105.
Type of Study: Case & Review |
Subject:
Pediatric Endocrinology
Received: 2018/02/15 | Accepted: 2018/05/30 | Published: 2019/04/1
Received: 2018/02/15 | Accepted: 2018/05/30 | Published: 2019/04/1
References
1. Khaetarpal P, Das Kh, Panigrahi I, Munshi A. Primoral dwarfism: Overview of clinical and genetic aspects. Molecular Genetics and Genomics. 2016; 291(1):1-15. [DOI:10.1007/s00438-015-1110-y] [PMID]) [DOI:10.1007/s00438-015-1110-y]
2. Codd PJ, Scott RM, Smith ER. Seckel syndrome and moyamoya: Case report. Journal of Neurosurgery: Pediatrics. 2009; 3(4):320-4. [DOI:10.3171/2008.12.PEDS08205] [PMID] [DOI:10.3171/2008.12.PEDS08205]
3. Klingsein A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Development. 2011; 25(19):2011-24. [DOI:10.1101/gad.169037] [PMID] [PMCID] [DOI:10.1101/gad.169037]
4. Filonava L, Torres AG, de Pouplana LR. A novel cause for primordial dwarfism revealed: Defective tRNA modification. Genome Biology. 2015; 16(1):216. [DOI:10.1186/s13059-015-0786-y] [PMID] [PMCID]
5. Vardhan BH, Muthu MS, Saraswathi K, Koteeswaran D. Bird-headed dwarf of Seckel. Journal of Indian Society of Pedodontics and Preventive Dentistry. 2007; 25(5):8-9.
6. Seckel HP, editor. Bird-headed dwarfs: Studies in developmental anthropology including human proportions. Pediatrics. 1961; 27(3):426-9.
7. Seymen F, Tuna b, Kayserili H. Seckel syndrome: Report accuses. Journal of Clinical Pediatric Dentistry. 2002; 26(3):305-9. [DOI:10.17796/jcpd.26.3.l02834m2827m0132] [PMID]
8. Kjær I, Hansen N, Becktor KB, Birkebaek N, Balslev T. Craniofacial morphology, dentition, and skeletal maturity in four siblings with seckel syndrome. The Cleft Palate-Craniofacial Journal. 2001; 38(6):645-51. [DOI:10.1597/1545-1569_2001_038_0645_cmdasm_2.0.co_2] [DOI:10.1597/1545-1569_2001_038_0645_cmdasm_2.0.co_2]
9. Mokrani Benhelli H, Gaillard L, Biasutto P, Guen T, Touzot F, Vasquez N, et al. Primary microcephaly, impaired DNA replication and genomic instability caused by compound heterozygous ATR mutations. Human Mutation. 2013; 34(2):374-84. [DOI:10.1002/humu.22245] [PMID] [DOI:10.1002/humu.22245]
10. Majweski F, Goecke T. Studies of microcephalic primordial dwarfism I: Approach to a delineation of the seckle syndrome. American Journal of Medical Genetics. 1982; 12(1):7-21. [DOI:10.1002/ajmg.1320120103] [PMID] [DOI:10.1002/ajmg.1320120103]
11. Meinecke P, Passarge E. Microcephalic osteodysplastic primordial dwarfism type I/II in sibs. Journal of Medical Genetics. 1991; 28(11):795-800. [DOI:10.1136/jmg.28.11.795] [DOI:10.1136/jmg.28.11.795]
12. Melinda J, Pierce RP, Morse RP. The neurologic findings in Taybi- Linder syndrome (MOPD I/II): Case report and review of the literature. American Journal of Medical genetics. 2012; 158(3):606-10. [DOI:10.1002/ajmg.a.33958] [PMID] [DOI:10.1002/ajmg.a.33958]
13. Rauch A. The shortest of the short: Pericentrin mutations and beyond. Best Practice & Research Clinical Endocrinology & Metabolism. 2011; 25(1):125-30. [DOI:10.1016/j.beem.2010.10.015] [PMID] [DOI:10.1016/j.beem.2010.10.015]
14. Hall JG, Flora C, Scott CI, Pauli RM, Tanaka KI. Majweski osteodysplastic primordial dwarfism type II (MOPD II): Natural history and clinical findings. American Journal of Medical Genetics. 2004; 130(1):55-72. [DOI:10.1002/ajmg.a.30203] [PMID] [DOI:10.1002/ajmg.a.30203]
15. Luck D, Robertson F, Ganesan V. Screening for cerebrovascular disease in Microcephalic Osteodyspalstic Primordial Dwarfism type II (MOPD II): An evidence- based proposal. Pediatric Neurology. 2013; 48(4):294-8. [DOI:10.1016/j.pediatrneurol.2012.12.010] [PMID] [DOI:10.1016/j.pediatrneurol.2012.12.010]
16. Kent T, Yamaguchi Jr, Jennifer B, Salam MA, Karen S, Myung MD, et al. Spinal deformity in Russell-silver syndrome. Spinal Deformity. 2015; 3(1):95-7. [DOI:10.1016/j.jspd.2014.06.003] [PMID] [DOI:10.1016/j.jspd.2014.06.003]
17. Rao VB, Lily K, Seema K, Ghosh K, Dipika M. Paternal reciprocal translocation t(11;16)(p13;q24.3) in a Silver–Russel syndrome patient. Annales de Génétique. 2003; 46(4):475-8. [DOI:10.1016/S0003-3995(03)00028-5] [DOI:10.1016/S0003-3995(03)00028-5]
18. Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu Amero SN, et al. Exploring the spectrum of 3‐M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clinical Endocrinology. 2012; 77(3):335-42. [DOI:10.1111/j.1365-2265.2012.04428.x] [DOI:10.1111/j.1365-2265.2012.04428.x]
19. Marsaud C, Rossingnol S, Tounian P, Netchine I, Dubern B. Prevalence and management of gastro intestinal manifestation in silver- Russell syndrome. Archives of Disease in Childhood. 2015; 100(4):353-8. [DOI:10.1136/archdischild-2013-305864] [PMID] [DOI:10.1136/archdischild-2013-305864]
20. Sonja A, De Munnik, Hoesloot E, Roukema J, Schoots J, Kneors N, et al. Meier- Gorlin syndrome. Orphanet Journal of Rare Diseases. 2015; 10:114. [DOI:10.1186/s13023-015-0322-x] [PMID] [PMCID] [DOI:10.1186/s13023-015-0322-x]
21. Shawky RM, Gamal R. Meier–Gorlin syndrome: An additional Egyptian patient with gastroesophageal reflux, hydronephrosis, renal stones and hypoplastic labia majora and minora with clitromegaly. Egyptian Journal of Medical Human Genetics. 2016; 17(4):397-400. [DOI:10.1016/j.ejmhg.2015.12.006]
22. Ranke MB, Lindberg A, Mullis PE, Geffner ME, Tanaka T, Cutfield WS, et al. Towards optimal treatment with growth hormone in short children and adolescents: Evidence and theses. Hormone Research in Pædiatrics. 2013; 79(2):51-67. [DOI:10.1159/000347121] [PMID] [DOI:10.1159/000347121]
23. Wakeling EL, Brioude F, Lokulo Sodipe O, O'connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nature Reviews Endocrinology. 2017; 13(2):105. [DOI:10.1038/nrendo.2016.138] [PMID]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
