
Sun, Jul 5, 2026


Volume 9, Issue 2 (4-2021)
J. Pediatr. Rev 2021, 9(2): 137-144 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Medghalchi A, Hassanzadeh Rad A, Dalili S. The Ophthalmological Manifestations of Various Inborn Errors of Metabolism: A Narrative Review. J. Pediatr. Rev 2021; 9 (2) :137-144
URL: http://jpr.mazums.ac.ir/article-1-351-en.html
URL: http://jpr.mazums.ac.ir/article-1-351-en.html



1- Eye Research Center, Guilan University of Medical Sciences, Rasht, Iran.
2- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran.
3- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran. ,setiladalili1346@yahoo.com
2- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran.
3- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran. ,
Full-Text [PDF 1528 kb]
(3832 Downloads)
| Abstract (HTML) (5233 Views)
Full-Text: (2766 Views)
1. Context
nborn errors of metabolism or Inherited Metabolic Disorders (IMD) are a class of genetic disorders that occur because of single-gene defects (1). It can be distinguished not only in neonates and infants but also in adults (2, 3). The diagnosis of IMD is challenging for physicians. Significant changes in the diagnosis, classification, and treatment of IMDs were improved by medical technologies and higher knowledge regarding the human genome. These changes help the physician to recognize and treat patients earlier. There exist various recognized IMDs; a number of them are reported in Iran as biotinidase deficiency, methylmalonic acidemia, and non-ketotic hyperglycinemia (4, 5, 6, 7). Early diagnosis, thorough laboratory assessments, and reference to tertiary care centers if needed are essential for these patients.
IMDs may induce small molecule diseases by affecting protein, carbohydrate, lipid, and nucleic acid. It can also be an organelle disease and impact lysosomes, mitochondria, peroxisomes, and cytoplasm. Besides, IMDs present diverse complications on the eyes, including the ocular abnormalities of conjunctiva, cornea, lens, retina, optic nerve, ocular motility or symmetrical bilateral involvement, as well as severe visual acuity in approximately 2-meter counting finger.
Despite extensive knowledge about the metabolic, biochemical, and molecular features of various IMDs, their exact pathogenesis requires further evaluation. Furthermore, assessing the effects of mechanisms contributing to systemic metabolic diseases to ocular defects needs to be clarified.
In this narrative review article, the authors searched Institute for Scientific Information (ISI), Web of Science, PubMed, and Google Scholar for the relevant evidence. We found several recent articles related to the effects of IMDs on the eyes. Authors collected and reported the overall ophthalmological problems consequent to the endocrinologists’ needs regarding this issue in conferences. Besides, this article summarized the effects of IMDs of amino acids, carbohydrate, lipid, glycoprotein, lysosomes, lipoprotein and lipid, and miscellaneous diseases, such as Wilson and Menkes diseases on eyes. A previous study reported a detailed article consisting of the pathophysiology and physiology of the disease and their influence on the eyes (8).
2. Results
Table 1 presents the classification of metabolic diseases affecting the eyes; the ocular manifestations of IMDs can be distinguished in different diseases.
.jpg)
Albinism: It is an inherited disorder of melanin synthesis. It has two types; the first type simultaneously affects the skin and eye (oculocutaneouso), and the second type only involves the eye (ocular). The ophthalmic manifestations of both types of albinism are refractive errors, reduced visual acuity, Iris transillumination defects due to decreased iris pigmentation, foveal hypoplasia, nystagmus, strabismus, albino fundus, and abnormal nerve fiber crossing (9).
Cystinosis: It is a metabolic disease that causes the accumulation of cystine in the cells. Its pathognomonic ophthalmic finding is the deposition of iridescent crystal in the peripheral cornea. The other ocular manifestations of cystinosis are the accumulation of crystals in the uvea, iris, ciliary body, and pigmentary retinopathy in the eye (10).
Homocystinuria: It occurs in the absence of cystathionine b-synthetase (the enzyme that converts homocysteine to cystathionine). The main ocular manifestation of homocystinuria is ectopia lentis. Lens subluxation is usually inferiorly; however, the luxation varies. The other ophthalmic manifestations are progressive myopia, increased intra ocular pressure, retinal detachment, and optic atrophy (11).
Hyperornithinemia (Gyrate Atrophy): It is autosomal recessive dystrophy due to the deficiency of the activity of the mitochondrial matrix enzyme, ornithine aminotransferase, and the elevation of ornithine in plasma. The main ocular finding is hyperpigmented fundi that separates with the islands of loss of retinal pigment epithelium and chorioretinal atrophy. The other eye feathers are myopia, night blindness, and cataract (12).
Figure 1 shows the conditions concerning amino acid metabolism in eyes.
.jpg)
Classic Galactosemia: It is the deficit in the activity of galactose-l-phosphate uridyl transferase. A cataract may occur as a result of the accumulation of galactitol, i.e. the end metabolite of galactose in the nucleus and deep cortex of the lens. It induces an oil drop appearance on retroillumination (13). Figure 2 shows the disorder induced by carbohydrate metabolism on the eyes.
.jpg)
Niemann-Pick: It is a rare genetic disease generated due to sphingomyelinase isoenzymes that affect the body’s ability to metabolize cellular cholesterol and lipids. There are 3 types of Niemann-Pick disease, including A, B, and C (14). There exist various eye involvement in Niemann-Pick disease.
Niemann-pick disease types A: It causes a cherry-red spot in the macula in 50% of infants. A brownish discoloration of the anterior lens capsule is another manifestation of type A.
Niemann-Pick Disease Type B: It is the mildest form of the disease. It causes a cherry-red spot in the macula, i.e. diagnostic.
Niemann-Pick Disease Type C: It has a vertical ophthalmoplegia and optic atrophy. There is no cherry-red spot present in this type (15).
Fabry disease: It is a rare inherited disease that prevents the body from making alpha-galactosidase A. The accumulation of ceramide triheoside was conducted on various tissues, such as blood vessels in the kidney, skin, gastrointestinal tract, central nervous system, heart, and eye. The relevant ophthalmic manifestations include increased vessel tortuosity and aneurysms in the conjunctiva, verticillata in the cornea, increased tortuosity of vessels in the retina, and faint spoke-like lines opacity at the posterior lens capsule in the lens (16).
Gaucher disease: It is the result of a buildup of certain fatty substances in specific organs, particularly the spleen and liver. It has 3 types; the ocular feature of Gaucher disease type 1 includes brownish pinguecula-like masses, containing Gaucher cells. In the retina, Gaucher cells can be observed as well. Gaucher disease, type 2 is the acute neuropathic infantile form that causes persistent retroflection of the head and signs of pseudobulbar palsy. The classic Gaucher triad consists of Trismus, Strabismus, and Opisthotonus. Gaucher disease, type 3 has manifests features, such as ocular apraxia and corneal opacification (17).
Metachromatic Leukodystrophy (MLD): Multiple Sulfatase Deficiency (MSD) is a rare form of late-infantile MLD. Its ophthalmologic features consist of skew deviation, optic atrophy, retinal degeneration, and cherry-red spot (18).
Gangliosidosis: It is caused by the accumulation of gangliosides and contains different types of lipid storage disorders. The cherry-red spot is the eye manifestation of it (19). Figure 3 shows the disorders induced by lipid metabolism on the eyes.
.jpg)
Ophthalmologic manifestations of type I (infantile) mannosidosis is lens opacity in the posterior cortex. Type II (juvenile) mannosidosis induces Punctate opacity throughout the lens. The strabismus, pallor, and blurring of the optic disc and retinal degeneration are other manifestations of mannosidosis (20).
Fucosidosis is a lysosomal storage disease caused by defective alpha-L-fucosidase with the accumulation of fucose in the tissues. Its ocular features include vascular tortuosities, tortuous conjunctival vessels with microaneurysms, and dilated and tortuous retinal veins (21). Figure 4 shows the disorders of glycoprotein
.jpg)
degradation.
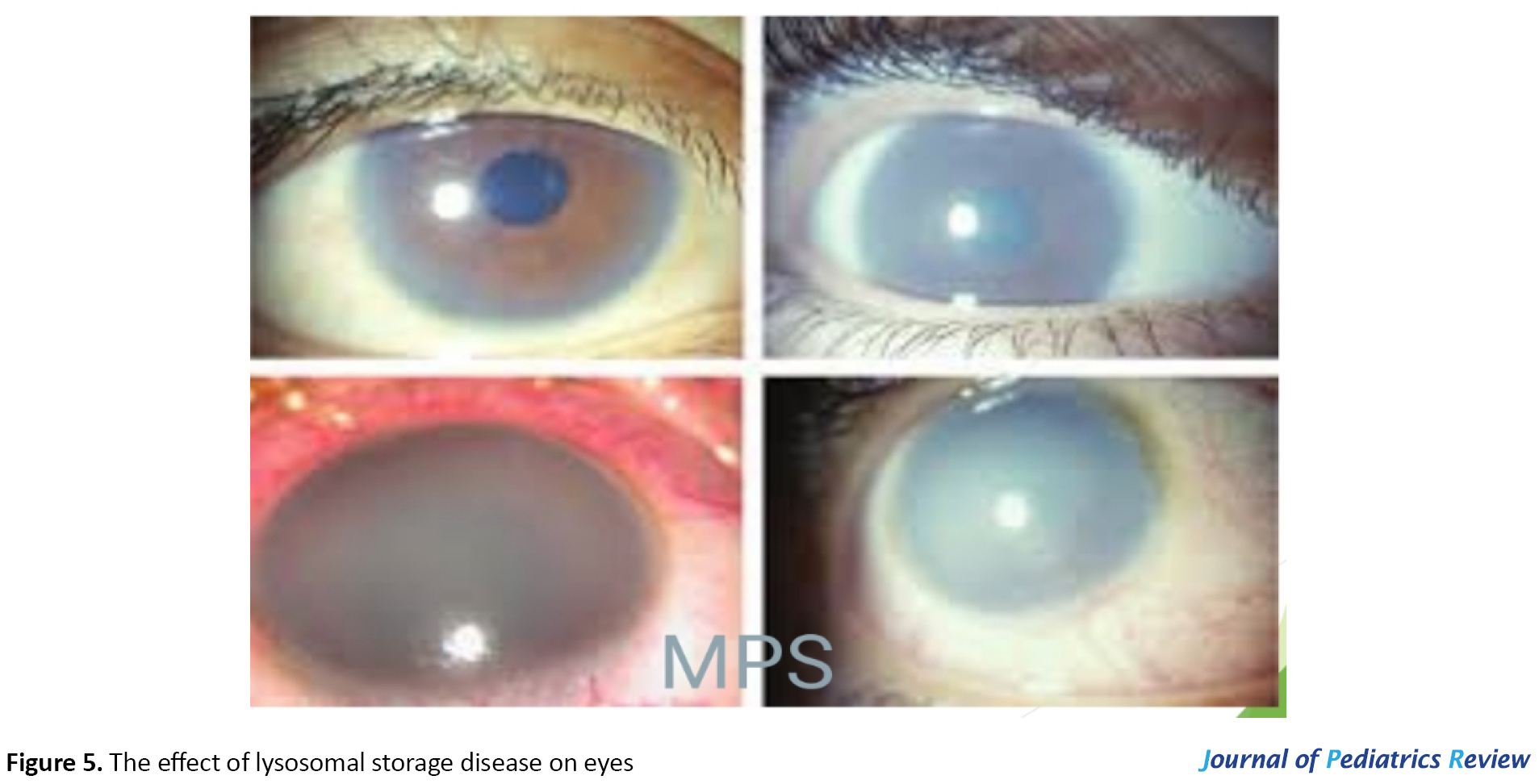
Lysosomal storage disease: Mucopolysaccharidosis (MPS) is a group of inherited lysosomal storage disorders. Their ocular manifestations are progressive corneal clouding, retinal pigmentary degeneration, diminished or extinguished electroretinogram, optic nerve head swelling, optic atrophy, and glaucoma (22). Figure 5 shows the effect of lysosomal storage disease on the eyes.
Wilson’s disease: It is an inherited disorder in which excess copper builds up in the liver, kidney, and basal ganglia of the brain. It induces the Kayser-Fleischer ring in the cornea and sunflower cataract. Kayser-Fleischer ring is determined by the band of golden to greenish-yellow, bronze, or brownish hue in the Descemet membrane of the peripheral of the cornea. Sunflower cataract occurs in only 15% to 20% of the affected individuals; it is determined by fine deposits beneath the anterior and posterior lens capsule, forming a disc-like opacity axially. The opacities do not interfere with vision (23).
Menkes disease: It is an X-linked recessive disorder of copper metabolism. The ocular manifestations of Menkes disease are hypopigmented fundus, a decrease in the nerve fibers of the optic nerve, an increase in the glial elements, and the attenuation or tortuosity of the retinal arterioles (24). Figure 6 shows the effect of miscellaneous disorders on the eyes.

3. Conclusion
Metabolic diseases are rare disorders, i.e. difficult to be diagnosed. They are usually difficult to be diagnosed based on the clinical findings, laboratory results, and physical examination. Besides, there is a shortage regarding the reports of numerous metabolic disorders in the worldwide genetic banks. Commonly, other organs should be assessed to accelerate the diagnosis. The eye is among the most significant organs, i.e. the mirror of the brain. In these disorders, ophthalmologists can help endocrinologists by thorough assessments of layers, including the sclera, cornea, pupil, and so on. Sometimes, metabolic diseases are asymptomatic; accordingly, there is no specific symptom or sign for the detection of those, leading clinicians to compile all assessments and examinations to reach a decision. Therefore, due to the direct toxic mechanisms of abnormal metabolites on eyes and regarding the effect of eye monitoring on the follow-up, management, and treatment of IMEs, a detailed ophthalmological assessment is essential.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflicts of interest
The authors declared no conflict of interest.
References
nborn errors of metabolism or Inherited Metabolic Disorders (IMD) are a class of genetic disorders that occur because of single-gene defects (1). It can be distinguished not only in neonates and infants but also in adults (2, 3). The diagnosis of IMD is challenging for physicians. Significant changes in the diagnosis, classification, and treatment of IMDs were improved by medical technologies and higher knowledge regarding the human genome. These changes help the physician to recognize and treat patients earlier. There exist various recognized IMDs; a number of them are reported in Iran as biotinidase deficiency, methylmalonic acidemia, and non-ketotic hyperglycinemia (4, 5, 6, 7). Early diagnosis, thorough laboratory assessments, and reference to tertiary care centers if needed are essential for these patients.
IMDs may induce small molecule diseases by affecting protein, carbohydrate, lipid, and nucleic acid. It can also be an organelle disease and impact lysosomes, mitochondria, peroxisomes, and cytoplasm. Besides, IMDs present diverse complications on the eyes, including the ocular abnormalities of conjunctiva, cornea, lens, retina, optic nerve, ocular motility or symmetrical bilateral involvement, as well as severe visual acuity in approximately 2-meter counting finger.
Despite extensive knowledge about the metabolic, biochemical, and molecular features of various IMDs, their exact pathogenesis requires further evaluation. Furthermore, assessing the effects of mechanisms contributing to systemic metabolic diseases to ocular defects needs to be clarified.
In this narrative review article, the authors searched Institute for Scientific Information (ISI), Web of Science, PubMed, and Google Scholar for the relevant evidence. We found several recent articles related to the effects of IMDs on the eyes. Authors collected and reported the overall ophthalmological problems consequent to the endocrinologists’ needs regarding this issue in conferences. Besides, this article summarized the effects of IMDs of amino acids, carbohydrate, lipid, glycoprotein, lysosomes, lipoprotein and lipid, and miscellaneous diseases, such as Wilson and Menkes diseases on eyes. A previous study reported a detailed article consisting of the pathophysiology and physiology of the disease and their influence on the eyes (8).
2. Results
Table 1 presents the classification of metabolic diseases affecting the eyes; the ocular manifestations of IMDs can be distinguished in different diseases.
Albinism: It is an inherited disorder of melanin synthesis. It has two types; the first type simultaneously affects the skin and eye (oculocutaneouso), and the second type only involves the eye (ocular). The ophthalmic manifestations of both types of albinism are refractive errors, reduced visual acuity, Iris transillumination defects due to decreased iris pigmentation, foveal hypoplasia, nystagmus, strabismus, albino fundus, and abnormal nerve fiber crossing (9).
Cystinosis: It is a metabolic disease that causes the accumulation of cystine in the cells. Its pathognomonic ophthalmic finding is the deposition of iridescent crystal in the peripheral cornea. The other ocular manifestations of cystinosis are the accumulation of crystals in the uvea, iris, ciliary body, and pigmentary retinopathy in the eye (10).
Homocystinuria: It occurs in the absence of cystathionine b-synthetase (the enzyme that converts homocysteine to cystathionine). The main ocular manifestation of homocystinuria is ectopia lentis. Lens subluxation is usually inferiorly; however, the luxation varies. The other ophthalmic manifestations are progressive myopia, increased intra ocular pressure, retinal detachment, and optic atrophy (11).
Hyperornithinemia (Gyrate Atrophy): It is autosomal recessive dystrophy due to the deficiency of the activity of the mitochondrial matrix enzyme, ornithine aminotransferase, and the elevation of ornithine in plasma. The main ocular finding is hyperpigmented fundi that separates with the islands of loss of retinal pigment epithelium and chorioretinal atrophy. The other eye feathers are myopia, night blindness, and cataract (12).
Figure 1 shows the conditions concerning amino acid metabolism in eyes.
Classic Galactosemia: It is the deficit in the activity of galactose-l-phosphate uridyl transferase. A cataract may occur as a result of the accumulation of galactitol, i.e. the end metabolite of galactose in the nucleus and deep cortex of the lens. It induces an oil drop appearance on retroillumination (13). Figure 2 shows the disorder induced by carbohydrate metabolism on the eyes.
Niemann-Pick: It is a rare genetic disease generated due to sphingomyelinase isoenzymes that affect the body’s ability to metabolize cellular cholesterol and lipids. There are 3 types of Niemann-Pick disease, including A, B, and C (14). There exist various eye involvement in Niemann-Pick disease.
Niemann-pick disease types A: It causes a cherry-red spot in the macula in 50% of infants. A brownish discoloration of the anterior lens capsule is another manifestation of type A.
Niemann-Pick Disease Type B: It is the mildest form of the disease. It causes a cherry-red spot in the macula, i.e. diagnostic.
Niemann-Pick Disease Type C: It has a vertical ophthalmoplegia and optic atrophy. There is no cherry-red spot present in this type (15).
Fabry disease: It is a rare inherited disease that prevents the body from making alpha-galactosidase A. The accumulation of ceramide triheoside was conducted on various tissues, such as blood vessels in the kidney, skin, gastrointestinal tract, central nervous system, heart, and eye. The relevant ophthalmic manifestations include increased vessel tortuosity and aneurysms in the conjunctiva, verticillata in the cornea, increased tortuosity of vessels in the retina, and faint spoke-like lines opacity at the posterior lens capsule in the lens (16).
Gaucher disease: It is the result of a buildup of certain fatty substances in specific organs, particularly the spleen and liver. It has 3 types; the ocular feature of Gaucher disease type 1 includes brownish pinguecula-like masses, containing Gaucher cells. In the retina, Gaucher cells can be observed as well. Gaucher disease, type 2 is the acute neuropathic infantile form that causes persistent retroflection of the head and signs of pseudobulbar palsy. The classic Gaucher triad consists of Trismus, Strabismus, and Opisthotonus. Gaucher disease, type 3 has manifests features, such as ocular apraxia and corneal opacification (17).
Metachromatic Leukodystrophy (MLD): Multiple Sulfatase Deficiency (MSD) is a rare form of late-infantile MLD. Its ophthalmologic features consist of skew deviation, optic atrophy, retinal degeneration, and cherry-red spot (18).
Gangliosidosis: It is caused by the accumulation of gangliosides and contains different types of lipid storage disorders. The cherry-red spot is the eye manifestation of it (19). Figure 3 shows the disorders induced by lipid metabolism on the eyes.
Ophthalmologic manifestations of type I (infantile) mannosidosis is lens opacity in the posterior cortex. Type II (juvenile) mannosidosis induces Punctate opacity throughout the lens. The strabismus, pallor, and blurring of the optic disc and retinal degeneration are other manifestations of mannosidosis (20).
Fucosidosis is a lysosomal storage disease caused by defective alpha-L-fucosidase with the accumulation of fucose in the tissues. Its ocular features include vascular tortuosities, tortuous conjunctival vessels with microaneurysms, and dilated and tortuous retinal veins (21). Figure 4 shows the disorders of glycoprotein
degradation.
Lysosomal storage disease: Mucopolysaccharidosis (MPS) is a group of inherited lysosomal storage disorders. Their ocular manifestations are progressive corneal clouding, retinal pigmentary degeneration, diminished or extinguished electroretinogram, optic nerve head swelling, optic atrophy, and glaucoma (22). Figure 5 shows the effect of lysosomal storage disease on the eyes.
Wilson’s disease: It is an inherited disorder in which excess copper builds up in the liver, kidney, and basal ganglia of the brain. It induces the Kayser-Fleischer ring in the cornea and sunflower cataract. Kayser-Fleischer ring is determined by the band of golden to greenish-yellow, bronze, or brownish hue in the Descemet membrane of the peripheral of the cornea. Sunflower cataract occurs in only 15% to 20% of the affected individuals; it is determined by fine deposits beneath the anterior and posterior lens capsule, forming a disc-like opacity axially. The opacities do not interfere with vision (23).
Menkes disease: It is an X-linked recessive disorder of copper metabolism. The ocular manifestations of Menkes disease are hypopigmented fundus, a decrease in the nerve fibers of the optic nerve, an increase in the glial elements, and the attenuation or tortuosity of the retinal arterioles (24). Figure 6 shows the effect of miscellaneous disorders on the eyes.
3. Conclusion
Metabolic diseases are rare disorders, i.e. difficult to be diagnosed. They are usually difficult to be diagnosed based on the clinical findings, laboratory results, and physical examination. Besides, there is a shortage regarding the reports of numerous metabolic disorders in the worldwide genetic banks. Commonly, other organs should be assessed to accelerate the diagnosis. The eye is among the most significant organs, i.e. the mirror of the brain. In these disorders, ophthalmologists can help endocrinologists by thorough assessments of layers, including the sclera, cornea, pupil, and so on. Sometimes, metabolic diseases are asymptomatic; accordingly, there is no specific symptom or sign for the detection of those, leading clinicians to compile all assessments and examinations to reach a decision. Therefore, due to the direct toxic mechanisms of abnormal metabolites on eyes and regarding the effect of eye monitoring on the follow-up, management, and treatment of IMEs, a detailed ophthalmological assessment is essential.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflicts of interest
The authors declared no conflict of interest.
References
- Korenev S, Lemonde H, Cleary M, Chakrapani A. Newborn screening for inborn errors of metabolism. Paediatrics and Child Health. 2019; 29(3):105-10. [DOI: 10.1016/j.paed.2019.01.002]
- El-Hattab AW. Inborn Errors of Metabolism. Clinics in Perinatology. 2015; 42(2):413-39. [DOI:10.1016/j.clp.2015.02.010] [PMID]
- Sedel F, Baumann N, Turpin JC, Lyon-Caen O, Saudubray JM, Cohen D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. Journal of Inherited Metabolic Disease. 2007; 30(5):631-41. [DOI:10.1007/s10545-007-0661-4] [PMID]
- Dalili S, Bidabadi E, Behnam B. Bell's palsy following growth hormone therapy in a patient with Prader-Willi syndrome: The first report. Genetics and Molecular Research. 2018; 17(1):gmr16039877. http://eprints.iums.ac.ir/7215/1/bells-palsy-following-growth-hormone-therapy-in-a-patient-with-praderwilli-syndrome-the-first-report.pdf
- Koohmanaee S, Zarkesh M, Tabrizi M, Rad Ah, Divshali S, Dalili S. Biotinidase deficiency in newborns as respiratory distress and tachypnea: A case report. Iranian Journal of Child Neurology. 2015; 9(2):58-60. [PMID] [PMCID]
- Karamizadeh Z, Dalili S, Karamifar H, Amirhakimi GH. Association of methylmalonic acidemia and erythema nodosum. Iranian Journal of Medical Sciences. 2011; 36(1):65-6. [PMID] [PMCID]
- Dalili S. Case presentation about non ketotic hyper glycinemia. Iranian Journal of Pediatrics. 2014; 24(suppl 2):S11. https://www.sid.ir/en/Journal/ViewPaper.aspx?ID=433668
- Medghalchi A, Hassanzadeh Rad A, Soltani-Moghadam R, Dalili S. The effect of amino acid, carbohydrate, and lipid metabolism disorders on eyes. Caspian Journal of Neurological Sciences. 2020; 6(3):190-16. [DOI:10.32598/CJNS.6.22.5]
- Williams R, Edwards M, Small DA, Edralin SK, Tornello I, Oxford J. Oculocutaneous Albinism in Embryonic Development [Internet]. 2020 [Updated April 22 2020]. Available from: https://www.boisestate.edu/undergraduate-research/2020/04/22/237-albinism-in-embryonic-development/
- Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: A review. Orphanet Journal of Rare Diseases. 2016; 11(1):47. [DOI:10.1186/s13023-016-0426-y] [PMID] [PMCID]
- Burke JP, O’Keefe M, Bowell R, Naughten ER. Ocular complications in homocystinuria: Early and late treated. British Journal of Ophthalmology. 1989; 73(6):427-31. [DOI:10.1136/bjo.73.6.427] [PMID] [PMCID]
- Kumar K, Agarwal A, Agarwal A, Dhawan A, Chandani N, Raj P. Ocular manifestations in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome: A rare association. Retinal Cases and Brief Reports. 2015; 9(2):134-7. [DOI:10.1097/ICB.0000000000000116] [PMID]
- Levy HL, Brown AE, Williams SE, de Juan Jr E. Vitreous hemorrhage as an ophthalmic complication of galactosemia. The Journal of Pediatrics. 1996; 129(6):922-5. [DOI:10.1016/S0022-3476(96)70041-0]
- Eskes EC, Sjouke B, Vaz FM, Goorden SM, van Kuilenburg AB, Aerts JM, et al. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Molecular Genetics and Metabolism. 2020; 130(1):16-26. [DOI:10.1016/j.ymgme.2020.02.002] [PMID]
- Rajappa M, Goyal A, Kaur J. Inherited metabolic disorders involving the eye: A clinico-biochemical perspective. Eye. 2010; 24(4):507-18. [DOI:10.1038/eye.2009.229] [PMID]
- Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S. Ocular manifestations of fabry’s disease: Data from the fabry outcome survey. British Journal of Ophthalmology. 2007; 91(2):210-4. [DOI:10.1136/bjo.2006.100602] [PMID] [PMCID]
- Michalski A, Leonard J, Taylor D. The eye and inherited metabolic disease: A review. Journal of the Royal Society of Medicine. 1988; 81(5):286-90. [DOI:10.1177/014107688808100517] [PMID] [PMCID]
- Bateman JB, Philippart M, Isenberg SJ. Ocular features of multiple sulfatase deficiency and a new variant of metachromatic leukodystrophy. Journal of Pediatric Ophthalmology and Strabismus. 1984; 21(4):133-9. [DOI:10.3928/0191-3913-19840701-04] [PMID]
- Biswas J, Nandi K, Sridharan S, Ranjan P. Ocular manifestation of storage diseases. Current Opinion in Ophthalmology. 2008; 19(6):507-11. [DOI:10.1097/ICU.0b013e32831215c3] [PMID]
- Cullen CL, Webb AA. Ocular manifestations of systemic disease. In: Veterinary Ophthalmology, 5th edition. Wiley Project: Gelatt Veterinary Ophthalmology Essentials of Veterinary Ophthalmology; 2008. https://www.researchgate.net/publication/327541078_Ocular_manifestations_of_systemic_disease
- Borrone C, Gatti R, Trias X, Durand P. Fucosidosis: Clinical, biochemical, immunologic, and genetic studies in two new cases. The Journal of Pediatrics. 1974; 84(5):727-30. [DOI:10.1016/S0022-3476(74)80019-3]
- Fenzl CR, Teramoto K, Moshirfar M. Ocular manifestations and management recommendations of lysosomal storage disorders I: Mucopolysaccharidoses. Clinical Ophthalmology. 2015; 9:1633-44. [DOI:10.2147/OPTH.S78368] [PMID] [PMCID]
- Tooley AA, Sweetser S. Clinical examination: Eyes. Clinical Liver Disease. 2016; 7(6):154-7. [DOI:10.1002/cld.561] [PMID] [PMCID]
- Jafri SK, Kumar R, Lashari SK, Chand P. Menkes disease: A rare disorder. The Journal of the Pakistan Medical Association. 2017; 67(10):1609-11. [PMID]
Type of Study: Narrative Review |
Subject:
Pediatric Endocrinology
Received: 2020/07/17 | Accepted: 2020/10/22 | Published: 2021/04/1
Received: 2020/07/17 | Accepted: 2020/10/22 | Published: 2021/04/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
