
Mon, Jul 7, 2025


Volume 9, Issue 2 (4-2021)
J. Pediatr. Rev 2021, 9(2): 83-96 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Mojibi N, Ghazanfari-Sarabi S, Hashemi-Soteh S M B. The Prevalence and Incidence of Congenital Phenylketonuria in 59 Countries: A Systematic Review. J. Pediatr. Rev 2021; 9 (2) :83-96
URL: http://jpr.mazums.ac.ir/article-1-352-en.html
URL: http://jpr.mazums.ac.ir/article-1-352-en.html



1- Department of Biochemistry and Biophysics, Faculty of Medicine, Mazandaran University of Medical Sciences, Sari, Iran.
2- Novin Genetics diagnostic laboratory, Farah Abad 4, Farah Abad Boulevard, Sari, Mazandaran, Iran
3- Immunogenetic Research Center, Molecular and Cell Biology Research Center, Faculty of Medicine, Mazandaran University of Medical Sciences, Sari, Iran. ,hashemisoteh@mazums.ac.ir
2- Novin Genetics diagnostic laboratory, Farah Abad 4, Farah Abad Boulevard, Sari, Mazandaran, Iran
3- Immunogenetic Research Center, Molecular and Cell Biology Research Center, Faculty of Medicine, Mazandaran University of Medical Sciences, Sari, Iran. ,
Full-Text [PDF 637 kb]
(3593 Downloads)
| Abstract (HTML) (6228 Views)
Full-Text: (4063 Views)
1. Context
Phenylalanine Hydroxylase (PAH; EC 1.14.16.1) is a hepatic enzyme, which hydroxylates the side-chain of Phenylalanine (Phe) to form Tyrosine (Tyr). Furthermore, the deficiency of PAH leads to Phenylketonuria (PKU, OMIM #261600), an autosomal recessive disorder (1). Besides, the PAH enzyme cofactor, tetrahydrobiopterin (BH4) decreases plasma Phe level in some patients with PKU. Therefore, the BH4 responsiveness must be monitored with a BH4 loading test. The 20%-29% reduction in blood Phe is defined as partial BH4 responsiveness; a >30% reduction is defined as the arbitrary BH4 responsiveness (2). There exists no consensus concerning PKU phenotype classification (3) due to the large range of cut-off points (4), and the time of neonatal screening. This is because patients often initiate the treatment before reaching the maximal Phe blood level (5). Moreover, Phe tolerance could not be applied to differentiate the PKU phenotypes. This is because due to the non-standard discrepancies and conditions between actual Phe that will be consumed and prescribed, it is controversial to determine the exact Phe tolerance. Recently, the PKU classification scheme has been simplified as follows: not requiring treatment; requiring diet, BH4, or both (6).
Most of the publications classified PKU based on its severity, which depends on the plasma Phe concentration; thus, we used the same previous scheme that categorized PKU in 3 classes, as follows: classic PKU (cPKU; Phe >1200 μmol/L), moderate PKU (modPKU; Phe 600-1200 μmol/L), and mild hyperphenylalaninemia (mHPA; 120-600 μmol/L) (7). The prompt diagnosis of PKU after birth can lead to the prescription of a low Phe diet, which can prevent the irreversible Intellectual Disability (ID). Although PKU screening in newborns is being performed in various developed countries, in Iran, it has been started in 2007 (8). PKU is the most frequent inborn error of amino acid metabolism affecting about 1 per 10000 Caucasians (9). Due to the approximate high rate of consanguineous marriage in developing countries, like Iran, it is predicted that the frequency of PKU will grow higher (10).
In case PKU patients remain undetected during the first week after birth, due to the increased level of Phe in the brain and blood, ID, motor deficits, autism, seizures, and microcephaly may be developed (3, 11). In this regard, it is of high importance to diagnose PKU shortly after birth. In this case, being equipped with an efficient screening program may prevent the development of PKU-induced complications in newborns. Therefore, throwing light on the frequency of PKU in various countries can raise awareness and lead to establishing an efficient NBS program. Therefore, it will be useful to recognize the most recent data on the worldwide prevalence of PKU as well as the prevalence of the disease in Iran; accordingly, these data could help to suggest new strategies for its prevention, diagnosis, and treatment. This systematic review explored the prevalence of PKU in different countries from January 2007 to December 2018. The relevant data could help to provide baseline information for the effective management of patients with PKU.
This systematic review was conducted based on the Preferred Reporting Items for Systematic Reviews and Meta-analysis (PRISMA) guidelines (12). Eligible literature was extracted from PubMed and ScienceDirect databases from January 2007 to December 2018 to identify studies reporting the global status of PKU prevalence.
2. Study Selection and data Extraction
Studies were screened using the following keywords: “Phenylketonuria”, “PKU”, “Phenylketonuria” or “PKU” and “Prevalence” or “Incidence” and “Iran” or “Middle East” or “Europe” or “America” or “Asia”. The exclusion criteria of the studies were as follows: studies not written in English; animals and in vitro studies; non-eligible publications, which included out-of-date data or PKU genotype frequency; duplicate reports, and studies presenting irrelevant data to PKU prevalence.
After a comprehensive search by two investigators (Sh. Ghazanfari, N. Mojibi) for PKU prevalence, 4306 (PubMed=1332, ScienceDirect=2974) relevant articles were identified, and 44 duplicates were removed. The relevance of remained 4262 studies was evaluated based on the titles/abstracts alone; of them, 223 studies were subjected to full-text review by authors based on the inclusion and exclusion criteria. Eventually, 44 of them were eligible to be included in the present systematic review (Figure 1).
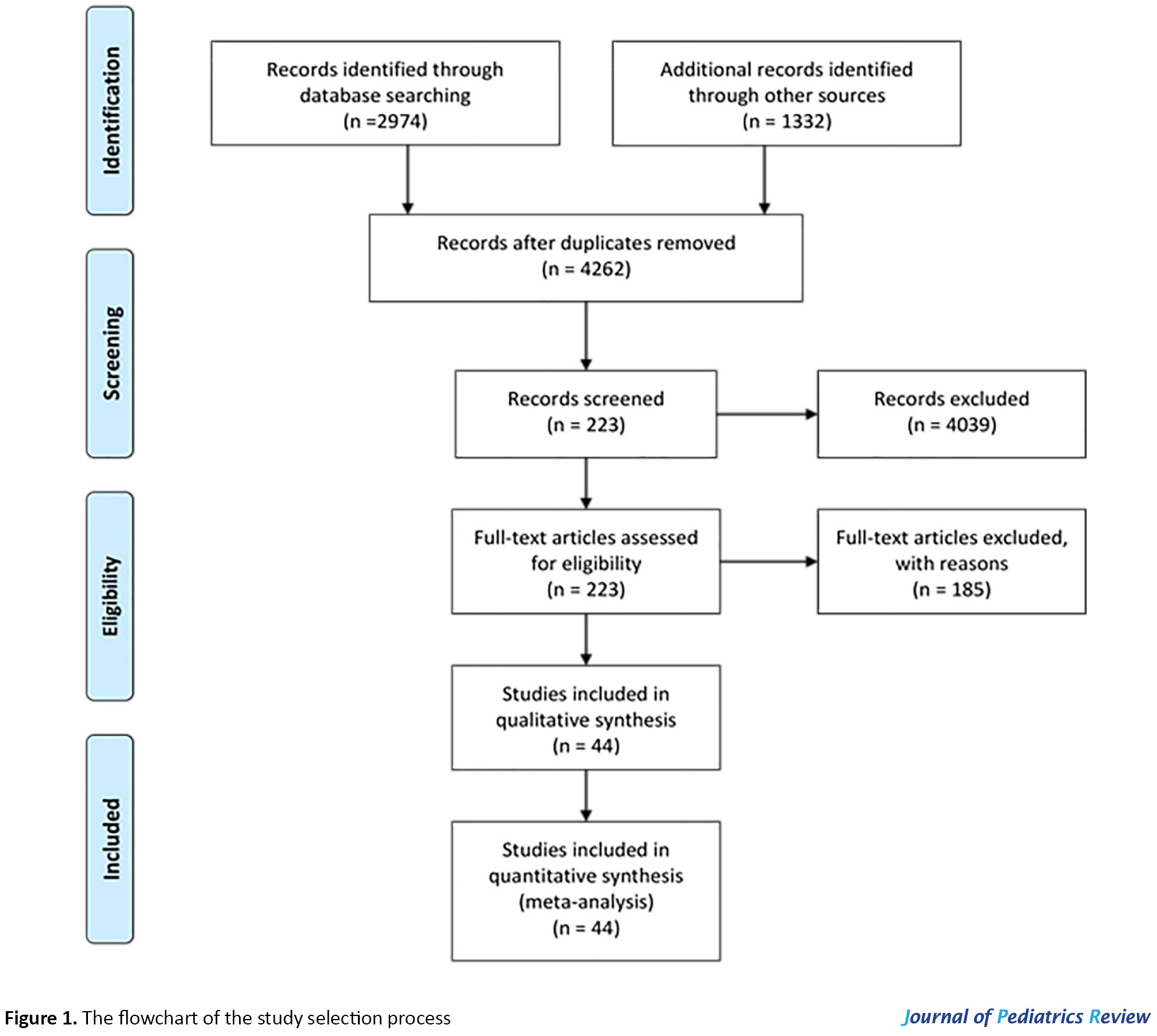
The references of the selected studies were also screened to obtain the relevant reports. Eventually, the authors independently evaluated selected studies to extract the required data. Any controversies were settled by consensus.
3. Results and Discussion
By searching the aforementioned databases, 4306 applicable articles were identified. Consequently, after removing 44 duplicates and 4039 non-eligible publications based on their language, the type of study, the date of publication, etc., the full texts and abstract of 223 publications were screened. Eventually, by the exclusion of 185 papers based on the inclusion/exclusion criteria, 44 publications were identified as qualified for the present systematic review (Figure 1).
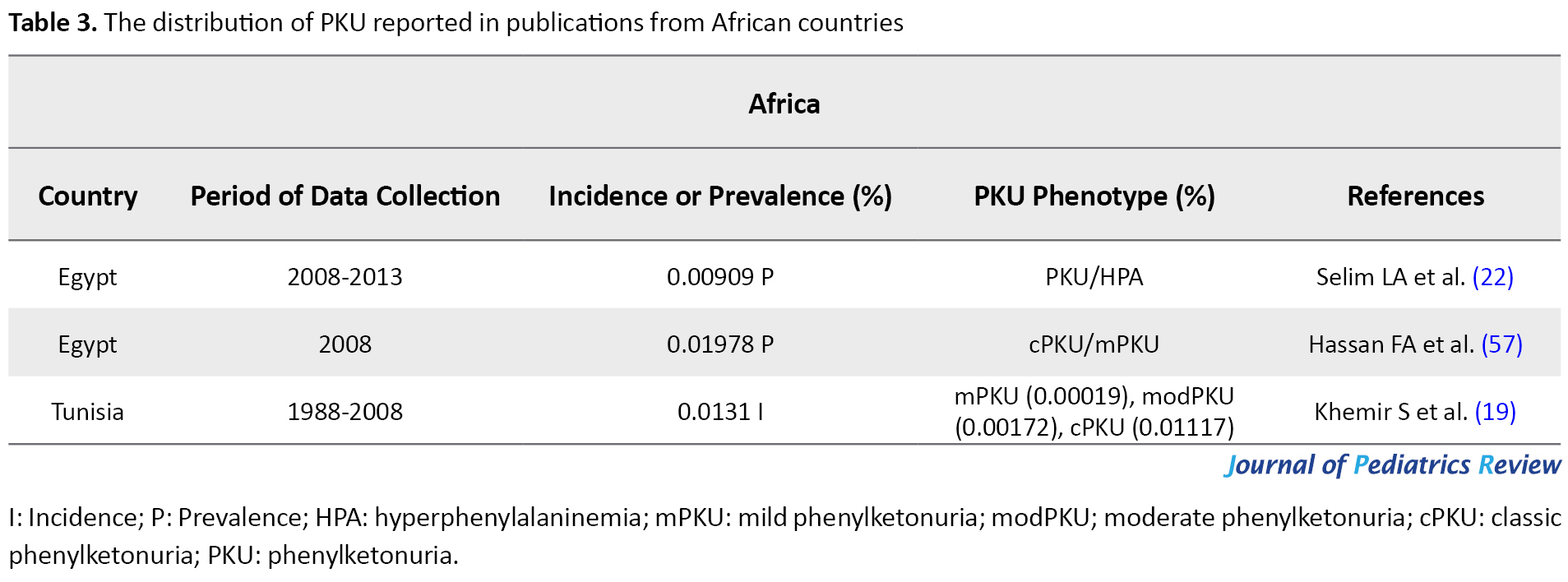
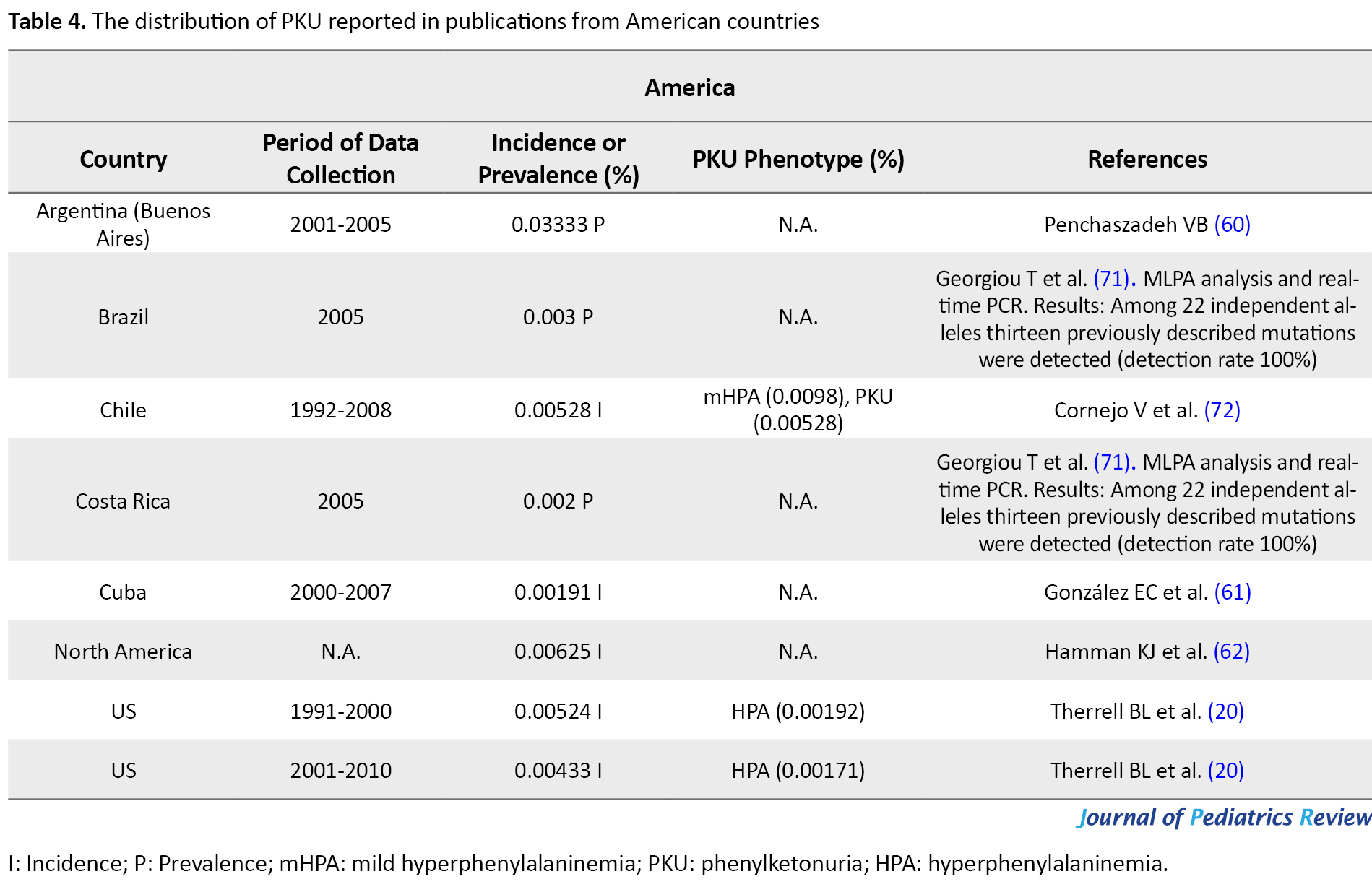
The flow chart of the selection process of eligible studies is shown in Figure 1. We identified 44 studies addressing the prevalence of PKU in 59 countries, including 15 Asian (Bahrain, China, India, Iran, Iraq, Japan, Korea, Lebanon, Saudi Arabia, Singapore, India, Taiwan, Thailand, Turkey, & United Arab Emirates; UAE), 36 European (Austria, Belarus, Belgium, Bosnia-Herzegovina, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Moldova, Netherlands, Norway, Poland, Portugal, Romania, Russia, Scotland, Serbia, Slovak Republic, Slovenia, Spain, Sweden, Switzerland, United Kingdom; UK, Ukraine, & Wales), two African (Egypt & Tunisia), and 6 American (the United States of America; U.S.A, Argentina, Brazil, Chile, Costa Rica, & Cuba) countries. The quantitative data derived from eligible studies are summarized in Tables 1, 2, 3 & 4.
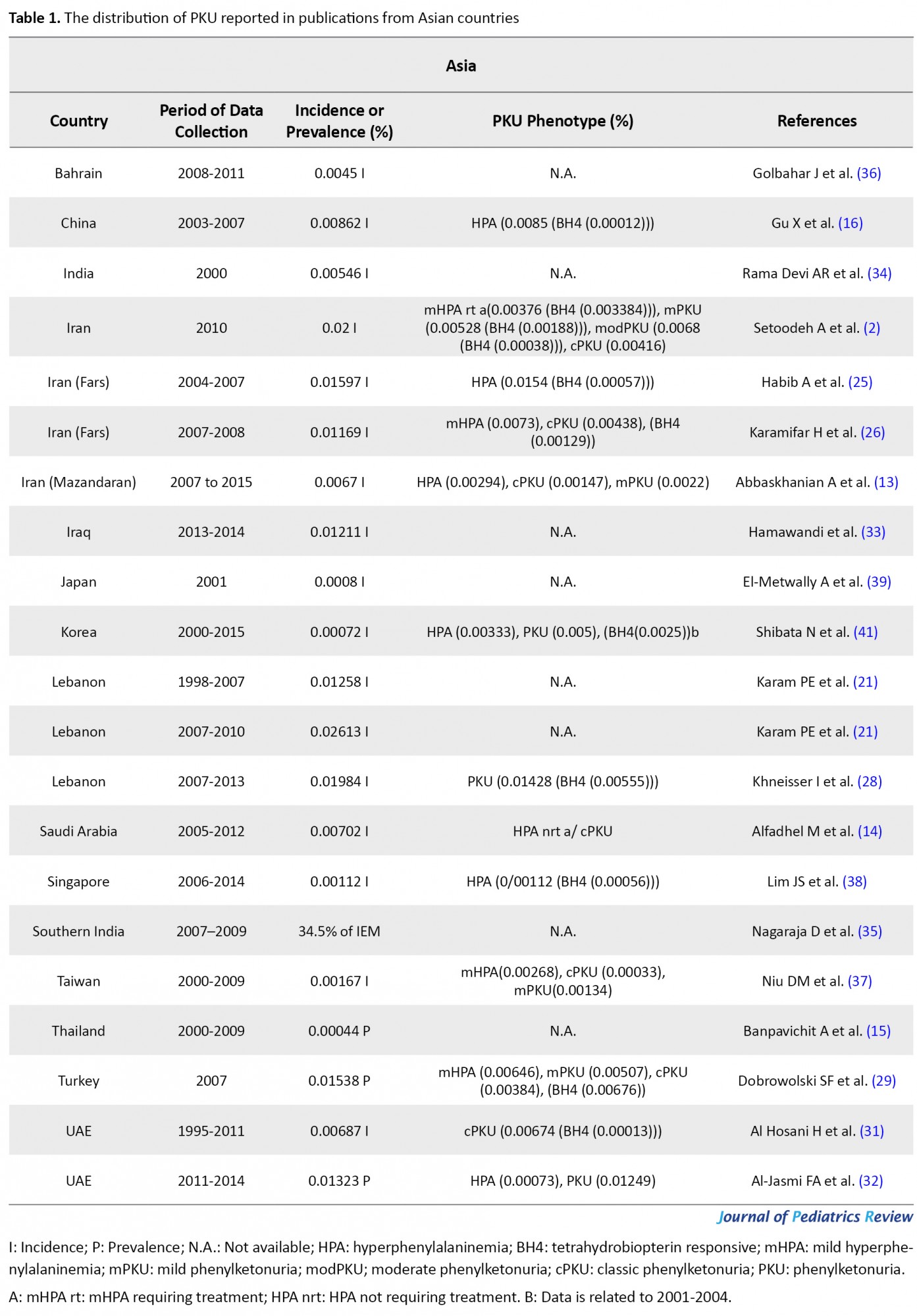
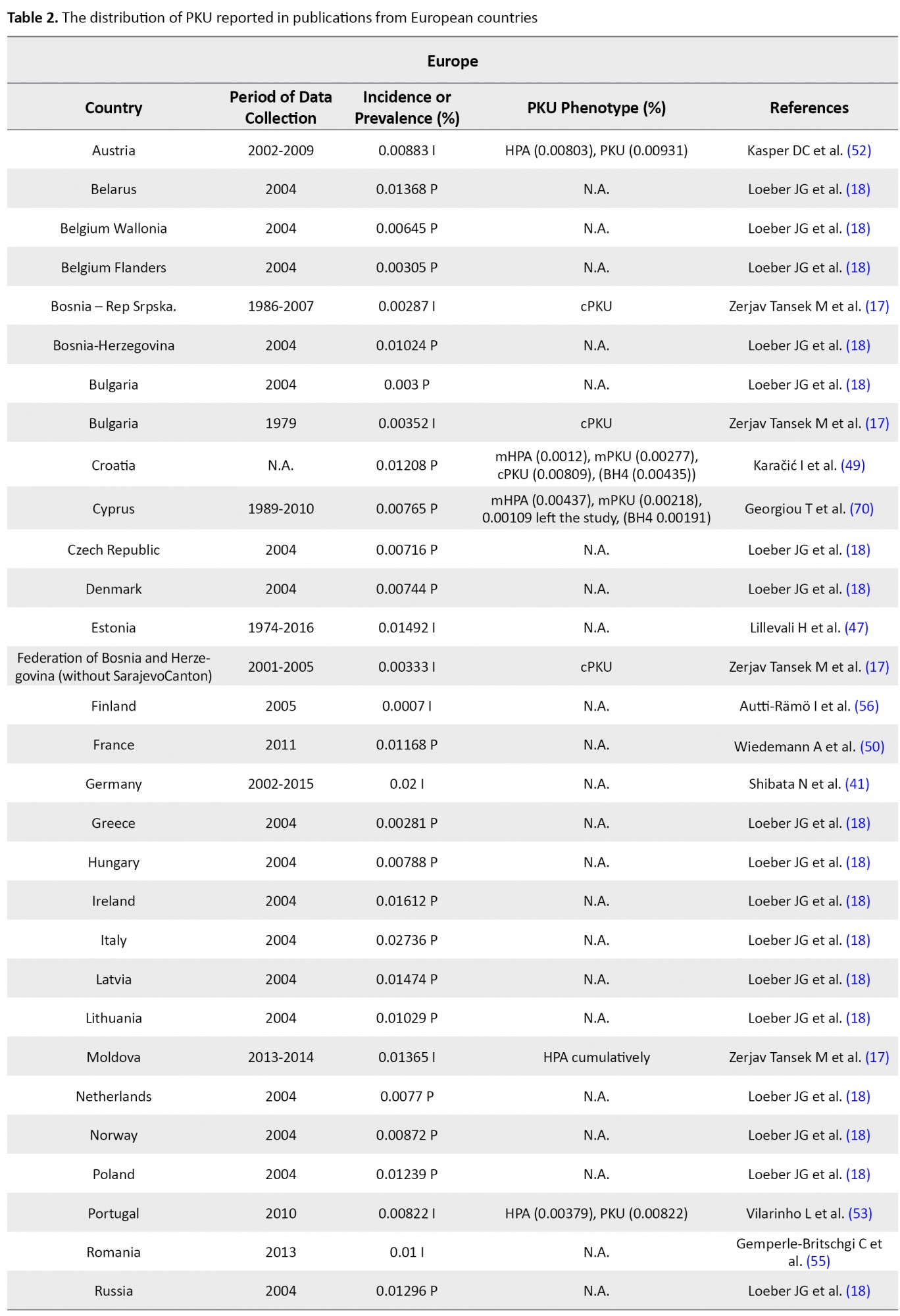
These studies were conducted based on recent surveys among opinion leaders in these 59 countries, i.e. in the majority members of the International Society for Neonatal Screening (ISNS). For Asian countries, such as Iran, Saudi Arabia, and Thailand, e.g., the reported data were provided by the Ministry of Health (13, 14, 15); in China, the same was reported by the National Neonatal Screening Quality Control, National Center for Clinical Laboratories (NCCL) (16) according to the national evaluation of PKU. Additionally, in Europe, previous international studies on the management of PKU mostly focused on the developed parts of Europe, while data about the large parts of southeastern and eastern Europe are scarce. In this regard, various eligible studies used in the present report, particularly the report by Zerjav Tansek et al. (2015), have meticulously covered PKU prevalence and incident in the southeastern Europe region (i.e. Balkan Peninsula) (17, 18).
In Tunisia, the related data were collected in association with the Association Tunisienne d'Etude des Maladies métaboliques héréditaires (ATEMMH), i.e. English is Tunisian association for the study of inherited metabolic diseases (19). Reported data for the U.S.A contributed to the Council of Regional Networks for Genetic Services (CORN) and the National Newborn Screening and Genetics Resource Center (now the National Newborn Screening & Global Resource Center; NNSGRC) reports through the National Newborn Screening Information System (NNSIS) (20). Although in other countries, like Lebonan NBS has not yet been covered by any national healthcare system, eligible tertiary care facilities, like various national and international universities. Concerning Lebonan, the American University of Beirut Medical Center (AUBMC), and Saint Joseph University (USJ) (21) or in Egypt, Cairo University Children's Hospital (CUCH) (22), are responsible for national biochemical genetic investigations.
The selected reports were published between January 2007 to December 2018. Ten studies had a case-control design, one was a cohort research, and the remainings were cross-sectional studies on PKU frequency. The age of the study participants ranged between 2 days and 11 years, with the sample sizes ranging from 3627 to 1100000 subjects. Except for 19 countries, which used tandem Mass Spectroscopy (MS/MS), 4 countries implemented bacterial inhibition assay. Besides, 4 studies applied High‐Pressure Liquid Chromatography (HPLC) and ELISA. Information on the applied methodology for 9 countries was unavailable; all other countries used a variety of the aforementioned methods to measure the blood Phe concentration.
Initially, unifying the calculation of prevalence rate per 100000 screened population and in the form of percentage was sought to obtain a dependable comparison of PKU prevalence/incidence. Then, studies were categorized via the population used to reckon PKU prevalence in national screening programs. The self-calculated prevalence/incidence of PKU (including BH4 dependent, classical type PKU, & HPA) was provided by the available information. The relevant extraction from included publications by dividing the number of cases by the sample size or the number of births from January 2007 to December 2018. Prevalence calculations were tabulated and expressed as the rate per 100000 of the screened population or as a percentage. We calculated PKU prevalence for all considered studies; however, only national programs yielded solid estimates. To report a reliable comparison, we converted PKU prevalence proportions, which in different studies were reported in various units, by percentage. Due to our calculation, Thailand presented the least percentage of PKU prevalence (0.00044%) and Italy faced the most prevalence of PKU, i.e. approximately 0.02736%. Moreover, the prevalence of PKU among patients with ID in Iran was reported by 2.1% in 2009.
A summary of the whole prevalence and incidence of PKU in numerous countries is presented in Tables 1, 2, 3 & 4. Most of the cases included in this study are not only cPKU, but also Hyperphenylalaninemia (HPA) not requiring treatment. Data related to all PKU variants frequency are listed in Tables 1, 2, 3 & 4; however, the relevant frequency of different PKU variants was not calculated in some of the references. Thus, we assumed the PKU frequency in these publications was related to all PKU phenotypes. In the case of the data availability related to the percentage of BH4 responsiveness among the different HPA and PKU variants in the eligible publications that were investigated for this study, this percentage is reported in Tables 1, 2, 3 & 4, as well. Extensive studies are available on the prevalence of PKU in Iran. The incidence of PKU in Iran in 1982 was estimated as 0.011% (23); however, this rate had raised to 0.02757% in 2002 (24), and it had later reduced to 0.02% of live births in 2015. Among these cases, 18.8%, 26.4%, 34%, and 20.8%, respectively presented mHPA (360-600 μmol/L blood Phe), mild PKU (mPKU; 600-900 μmol/L blood Phe), modPKU (900-1200 μmol/L blood Phe), and cPKU (blood Phe >1200 μmol/L) (2). Using HPLC in Fars Province, the south of Iran, the incidence of PKU from 2004 to 2007 was reported to be 0.01597% (25); however, this proportion reduced to 0.01169% from 2007 to -2008 by the screening of a total of 76966 newborns (62.5% mHPA &d 37.5% PKU) (26).
In a descriptive retrospective study in Mazandaran Province, northern Iran, 0.00662% of newborns were diagnosed with PKU from 2007 to 2015 using ELISA and HPLC as the confirmation methods. HPA phenotypes in Mazandaran was reported as 22.2% for cPKU, 33.3% for mPKU, and 44.5% for HPA (13). Another study in patients with ID (except Down syndrome) in Tehran and 31 other cities of Iran exhibited the %2.1 prevalence of cPKU and %0.44 of mHPA cases (27). These studies revealed that despite the implementation of the Newborn Screening (NBS) programs for an Inborn Error of Metabolism (IEM) in most cities of Iran, the prevalence of PKU remained significant.
The frequency of the disease in Asian countries
According to our findings, based on the Ministry of Health, PKU was highly prevalent in Iran (0.02%), particularly in Fars Province with a prevalence of 0.01597% from 2004 to 2007 (25). This rate was respectively higher than Turkey, UAE, Lebanon, Iraq, China, Saudi Arabia, Mazandaran (Iran), India, Bahrain, Taiwan, Singapore, Japan, Korea, and Thailand. Nevertheless, PKU occurrence experienced a reduction to 0.01169% in Fras Province in the following years (2007-2008) (26), i.e. less than PKU incidence in Iraq, Lebanon, UAE, and Turkey. Although in Lebanon from 1998 to 2007, only 18 cases of PKU variants were detected by screening 143000 newborns (0.01258%); after introducing expanded NBS by MS/MS in 2007, the incidence of PKU equaled 0.02613%. This rate reduced to 0.01984% of newborns by 2013 (21, 28); the PKU disorders were of the highest prevalence in this country among the other Asian countries. After Lebanon and Iran, the highest PKU prevalence belonged to Turkey with 0.01538% of newborns in 2007 (29). In the UAE however, the incidence of PKU from 1998 to 2000 was nearly close to Thailand, which had raised from 0.00498% (30) to 0.00687% (89% cPKU & 1.75% PKU) in 1995-2011 (31). PKU prevalence reached 0.01323 % (only cPKU) in 2011-2014; UAE was ranked fourth for having a high prevalence of PKU in the investigated Asian countries (Table 1) (32). Besides, 0.01211% of newborns presented PKU in Sulaimani City in Iraq. Accordingly, Hamawandi et al. claimed that using Iranian kit materials and lab equipment, as well as the similar ethnicity of Fars and Kurdish population might be the possible explanations for this similarity (33).
The rate of PKU incidence in China and Saudi Arabia were more than in Mazandaran Province in Iran and measured as 0.00862% (16) and 0.00702% (14), respectively. In some countries, we only could find outdated data, e.g., the latest PKU incidence report in India was published in 2004 (0.00546%) (34); however, this rate in Southern India was reported as 34.5% of IEM in 2007-2009 (35). The United Kingdom of Bahrain reported the PKU incidence of 0.0045% (36). In Taiwan, from 2001 to 2014, 0.00167% of Taiwanese live births were detected with PKU (37). An 8-year study in Singapore revealed an incidence of 0.00112% for PKU (38). This rate was 0.0008% in Nagasaki, Japan, in 2001 (39). The reason for the low incidence of PKU in Japan was claimed to be because of genetic drift in the founder population of Japanese islands (40). In South Korea, NBS on 3.44 million in 2000-2015 indicated 0.00072% of newborns with PKU (41). Among Asian countries, Thailand possessed the least prevalence of PKU (0.00044%) (15) after Korea. We can suggest the high rate of the heterogeneity of PAH locus (42, 43) and consanguineous marriages among the Iranian and Turkish population (10, 27, 44) as remarkable causes of the high occurrence of PKU; thus, these countries are of high rank in PKU frequency in Asia.
The frequency of the disease in European countries
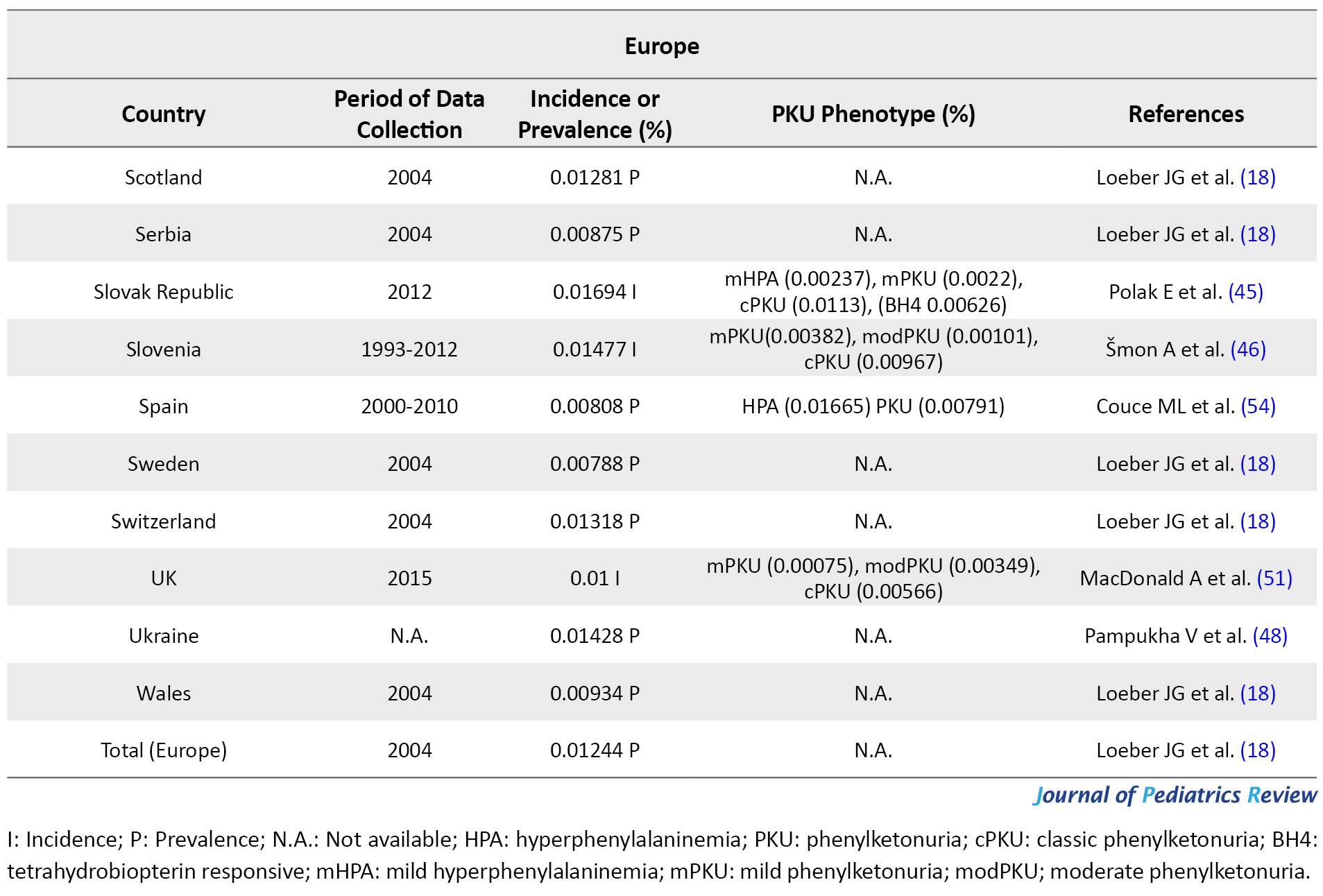
In Europe, apart from Malta and Finland (because of their known low PKU incidence), all countries use a national program for PKU; although in some countries, these programs fail to have 100% coverage. Italy (0.02736%) (18), and Germany (0.02%) (41) presented the highest frequency of PKU among European countries. Slovak Republic (Bratislava, Banska Bystrica, & Kosice centers) in 2012 and Ireland in 2004 with 0.01694% PKU incidence (45, 46) and 0.01612% PKU prevalence (18) detected among newborns, respectively possessed the highest PKU frequency after Germany and Italy. The incidence of PKU had an estimated 0.01492% of newborns in Estonia from 1974 to 2016 (47), i.e. almost close to Slovenia (0.01477%, cPKU=0.0098%) (46) and PKU prevalence in Latvia (0.01474%) (18). However, the ethnic origin of the reported subjects in Estonia was 26% of Slavic (Russian), 7% of mixed-origin, and the rest were Estonian. Furthermore, as per the State Expert Center MoH, PKU prevalence was reported to be 0.01428% in Ukrainians (48). Moreover, PKU prevalence was documented as 0.01318% for Switzerland (18). This rate is almost close to PKU frequency in Russia (0.01296%), Scotland (0.01281%), Poland (0.01239%), and Croatia (0.01208%; 10% of the patients were classified as mHPA, 23% were assigned to mPKU, and 67% to cPKU) (18, 49).
In other European countries, PKU frequency was reported as 0.01168% in France (50), 0.01% in UK (51), 0.00833% in Austria (52), 0.00872% in Norway (18), 0.00822% in 2010 in Portugal (53), 0.00808% in Spain (HPA=0.016%) (54), 0.00788% in Sweden (18), 0.00788% in Hungary, 0.0077% in Netherlands (18), and 0.00744 in Denmark (18). Although the official incidence of PKU in Romania remains unpublished, it has been reported by Bucharest National Authority for neonatal screening diagnostic confirmation and specific treatment in PKU and congenital hypothyroidism as 0.01% of live births in 2013 (55). PKU incidence in some other European countries is summarized in Table 2. Among European countries, Finland experienced the lowest PKU frequency by an incidence of 0.001-0.0005; however, this is the latest incidence we could find in our database, i.e. related to 2005 (56). Due to our investigation, the highest and lowest rate of PKU distribution belonged to Italy (0.027%) and Finland (0.0007%), respectively. Notably, the incidence of PKU in Germany was respectively 25- and 45-fold higher than that in Japan and Thailand; however, it was equal to the occurrence of this disorder in Iran.
The frequency of the disease in African countries
The prevalence of PKU in Egypt was evaluated as 0.01978% of infants in 2008 (57), by 2013 this rate reduced to 0.00909%. In 2012, PKU was the most common aminoacidopathies disorder among Tunisians as another African country (58). Khemir et al. estimated the incidence of PKU in Tunisia as 0.0131% from 1988 to 2008 (19). Insufficient diagnostic capabilities in Africa might obstruct revealing the actual rate of PKU frequency; in general, the occurrence of PKU has been reported as low in Africa (59). In this regard, the high frequency of PKU in the north of Africa might be owed to the high level of consanguineous marriage (Table 3).
The frequency of the disease in American countries
In 2005, Buenos Aires in Argentina (0.03333%) (60) possessed the highest, and Cuba (0.00191) (61) had the lowest rate of PKU incidence among Latin American countries. Based on the calculated occurrence of IEM in the U.S.A through NBS by the National Newborn Screening Information System (NNSIS), the US witnessed a decrease in PKU incidence from 0.00524% in 1991-2000 to 0.00433% in 2001-2010 (20). By 2014, 10000 individuals in the US were diagnosed with PKU. In 2001-2010, Hawaii was the US state with the least prevalence of PKU (0.00054%) and Wyoming presented the most PKU prevalence (0.016%) (20). In northern America, the PKU incidence rate was reported as 0.00625% (62). Moreover, PKU occurs in 0.01% of Caucasians (9) and 0.00191% of African Americans in the US (63) (Table 4).
Certain restrictions have affected this review. First, the main search was limited to the publications in English. A major disadvantage of this review concerned its design, in particular for prevalence/incidence studies in which data from most of the reports are based on the collection of the retrospective data either from registries or medical records. Such routine data possess some pitfalls, such as imprecision or deficiency. Another possible limitation included the small sample sizes. Other limitations involved the method for calculating PKU prevalence/incidence in the reports. Some reports applied the divisor as several whole live births and not the actual number of screened cases; however, others performed PKU prevalence/incidence evaluation using a total number of abnormal cases instead of total screened subjects. However, in the case of these reports using self-calculations, we tried to provide more precise data of PKU prevalence/incidence.
4. Conclusion
Advances in science and technology drive the evolution of methods, which allow the accurate measurements of acylcarnitines, amino acids, and other critical metabolites in the diagnosis of metabolic disorders, like MS/MS (64). Due to its sensitivity, selectivity, the ability to screen several diseases simultaneously, and short-term analysis, MS/MS has been widely used for NBS (65). Moreover, the widespread use of MS/MS can significantly be contributed to the estimating of the frequency of incidence of each IEM in our population; reports from countries that have incorporated NBS indicated that the occurrence of some disorders is higher than expected (38, 66). Overall, the prevalence of PKU worldwide in 2015 was claimed to be 0.00641% of live births (67). This review aimed to report the PKU prevalence in different countries to raise awareness and leads to establishing an efficient NBS program. Surprisingly, the highest and lowest relevant rates belonged to Asia and a European country, respectively. Therefore, with a slight difference between Iran, Germany, and Italy, the highest rate of incidence of PKU belonged to Italy with approximately 0.02736% of total live newborns. In contrast, the lowest rate of PKU distribution in Asia belonged to Thailand (about 0.0004%), and in Europe, it concerned Finland. Although we could not conclude the prevalence of PKU in Africa just based on two studies, this prevalence in Tunisia was about the same as some European and Asian countries. In conclusion, in the investigated studies, PKU had covered more than 50% of IEMs in Germany and Italy (68, 69), while this rate for Asian countries (Oman, Japan, Bahrain, India, China) ranged between 6% and 20% of IEMs (data not presented) (70). These findings indicated that the incidence rates of amino acids differ between European and Asian populations.
Our research demonstrated the necessity of conducting further investigations on PKU prevalence, especially in Iran and other countries in the Middle East. Thus, the priority is to assist the countries where the basic screening programs encompass less than 100% coverage. Additionally, continuous monitoring of the NBS program could help to decrease the variation in design and methodology using the available knowledge and expertise in the literature. The considerable difference in recalling the PKU incidence rates represents one important and obvious area for the refinement of NBS program performances, globally. Since treatment depends on the early diagnosis of the disorder, the implementation of neonatal screening for PKU is crucial. Moreover, the estimation of the exact prevalence of the disorder is required to implement effective policies and strategies for controlling the disease occurrence.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflicts of interest
The authors declared no conflicts of interest.
References
Phenylalanine Hydroxylase (PAH; EC 1.14.16.1) is a hepatic enzyme, which hydroxylates the side-chain of Phenylalanine (Phe) to form Tyrosine (Tyr). Furthermore, the deficiency of PAH leads to Phenylketonuria (PKU, OMIM #261600), an autosomal recessive disorder (1). Besides, the PAH enzyme cofactor, tetrahydrobiopterin (BH4) decreases plasma Phe level in some patients with PKU. Therefore, the BH4 responsiveness must be monitored with a BH4 loading test. The 20%-29% reduction in blood Phe is defined as partial BH4 responsiveness; a >30% reduction is defined as the arbitrary BH4 responsiveness (2). There exists no consensus concerning PKU phenotype classification (3) due to the large range of cut-off points (4), and the time of neonatal screening. This is because patients often initiate the treatment before reaching the maximal Phe blood level (5). Moreover, Phe tolerance could not be applied to differentiate the PKU phenotypes. This is because due to the non-standard discrepancies and conditions between actual Phe that will be consumed and prescribed, it is controversial to determine the exact Phe tolerance. Recently, the PKU classification scheme has been simplified as follows: not requiring treatment; requiring diet, BH4, or both (6).
Most of the publications classified PKU based on its severity, which depends on the plasma Phe concentration; thus, we used the same previous scheme that categorized PKU in 3 classes, as follows: classic PKU (cPKU; Phe >1200 μmol/L), moderate PKU (modPKU; Phe 600-1200 μmol/L), and mild hyperphenylalaninemia (mHPA; 120-600 μmol/L) (7). The prompt diagnosis of PKU after birth can lead to the prescription of a low Phe diet, which can prevent the irreversible Intellectual Disability (ID). Although PKU screening in newborns is being performed in various developed countries, in Iran, it has been started in 2007 (8). PKU is the most frequent inborn error of amino acid metabolism affecting about 1 per 10000 Caucasians (9). Due to the approximate high rate of consanguineous marriage in developing countries, like Iran, it is predicted that the frequency of PKU will grow higher (10).
In case PKU patients remain undetected during the first week after birth, due to the increased level of Phe in the brain and blood, ID, motor deficits, autism, seizures, and microcephaly may be developed (3, 11). In this regard, it is of high importance to diagnose PKU shortly after birth. In this case, being equipped with an efficient screening program may prevent the development of PKU-induced complications in newborns. Therefore, throwing light on the frequency of PKU in various countries can raise awareness and lead to establishing an efficient NBS program. Therefore, it will be useful to recognize the most recent data on the worldwide prevalence of PKU as well as the prevalence of the disease in Iran; accordingly, these data could help to suggest new strategies for its prevention, diagnosis, and treatment. This systematic review explored the prevalence of PKU in different countries from January 2007 to December 2018. The relevant data could help to provide baseline information for the effective management of patients with PKU.
This systematic review was conducted based on the Preferred Reporting Items for Systematic Reviews and Meta-analysis (PRISMA) guidelines (12). Eligible literature was extracted from PubMed and ScienceDirect databases from January 2007 to December 2018 to identify studies reporting the global status of PKU prevalence.
2. Study Selection and data Extraction
Studies were screened using the following keywords: “Phenylketonuria”, “PKU”, “Phenylketonuria” or “PKU” and “Prevalence” or “Incidence” and “Iran” or “Middle East” or “Europe” or “America” or “Asia”. The exclusion criteria of the studies were as follows: studies not written in English; animals and in vitro studies; non-eligible publications, which included out-of-date data or PKU genotype frequency; duplicate reports, and studies presenting irrelevant data to PKU prevalence.
After a comprehensive search by two investigators (Sh. Ghazanfari, N. Mojibi) for PKU prevalence, 4306 (PubMed=1332, ScienceDirect=2974) relevant articles were identified, and 44 duplicates were removed. The relevance of remained 4262 studies was evaluated based on the titles/abstracts alone; of them, 223 studies were subjected to full-text review by authors based on the inclusion and exclusion criteria. Eventually, 44 of them were eligible to be included in the present systematic review (Figure 1).
The references of the selected studies were also screened to obtain the relevant reports. Eventually, the authors independently evaluated selected studies to extract the required data. Any controversies were settled by consensus.
3. Results and Discussion
By searching the aforementioned databases, 4306 applicable articles were identified. Consequently, after removing 44 duplicates and 4039 non-eligible publications based on their language, the type of study, the date of publication, etc., the full texts and abstract of 223 publications were screened. Eventually, by the exclusion of 185 papers based on the inclusion/exclusion criteria, 44 publications were identified as qualified for the present systematic review (Figure 1).
The flow chart of the selection process of eligible studies is shown in Figure 1. We identified 44 studies addressing the prevalence of PKU in 59 countries, including 15 Asian (Bahrain, China, India, Iran, Iraq, Japan, Korea, Lebanon, Saudi Arabia, Singapore, India, Taiwan, Thailand, Turkey, & United Arab Emirates; UAE), 36 European (Austria, Belarus, Belgium, Bosnia-Herzegovina, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Moldova, Netherlands, Norway, Poland, Portugal, Romania, Russia, Scotland, Serbia, Slovak Republic, Slovenia, Spain, Sweden, Switzerland, United Kingdom; UK, Ukraine, & Wales), two African (Egypt & Tunisia), and 6 American (the United States of America; U.S.A, Argentina, Brazil, Chile, Costa Rica, & Cuba) countries. The quantitative data derived from eligible studies are summarized in Tables 1, 2, 3 & 4.
These studies were conducted based on recent surveys among opinion leaders in these 59 countries, i.e. in the majority members of the International Society for Neonatal Screening (ISNS). For Asian countries, such as Iran, Saudi Arabia, and Thailand, e.g., the reported data were provided by the Ministry of Health (13, 14, 15); in China, the same was reported by the National Neonatal Screening Quality Control, National Center for Clinical Laboratories (NCCL) (16) according to the national evaluation of PKU. Additionally, in Europe, previous international studies on the management of PKU mostly focused on the developed parts of Europe, while data about the large parts of southeastern and eastern Europe are scarce. In this regard, various eligible studies used in the present report, particularly the report by Zerjav Tansek et al. (2015), have meticulously covered PKU prevalence and incident in the southeastern Europe region (i.e. Balkan Peninsula) (17, 18).
In Tunisia, the related data were collected in association with the Association Tunisienne d'Etude des Maladies métaboliques héréditaires (ATEMMH), i.e. English is Tunisian association for the study of inherited metabolic diseases (19). Reported data for the U.S.A contributed to the Council of Regional Networks for Genetic Services (CORN) and the National Newborn Screening and Genetics Resource Center (now the National Newborn Screening & Global Resource Center; NNSGRC) reports through the National Newborn Screening Information System (NNSIS) (20). Although in other countries, like Lebonan NBS has not yet been covered by any national healthcare system, eligible tertiary care facilities, like various national and international universities. Concerning Lebonan, the American University of Beirut Medical Center (AUBMC), and Saint Joseph University (USJ) (21) or in Egypt, Cairo University Children's Hospital (CUCH) (22), are responsible for national biochemical genetic investigations.
The selected reports were published between January 2007 to December 2018. Ten studies had a case-control design, one was a cohort research, and the remainings were cross-sectional studies on PKU frequency. The age of the study participants ranged between 2 days and 11 years, with the sample sizes ranging from 3627 to 1100000 subjects. Except for 19 countries, which used tandem Mass Spectroscopy (MS/MS), 4 countries implemented bacterial inhibition assay. Besides, 4 studies applied High‐Pressure Liquid Chromatography (HPLC) and ELISA. Information on the applied methodology for 9 countries was unavailable; all other countries used a variety of the aforementioned methods to measure the blood Phe concentration.
Initially, unifying the calculation of prevalence rate per 100000 screened population and in the form of percentage was sought to obtain a dependable comparison of PKU prevalence/incidence. Then, studies were categorized via the population used to reckon PKU prevalence in national screening programs. The self-calculated prevalence/incidence of PKU (including BH4 dependent, classical type PKU, & HPA) was provided by the available information. The relevant extraction from included publications by dividing the number of cases by the sample size or the number of births from January 2007 to December 2018. Prevalence calculations were tabulated and expressed as the rate per 100000 of the screened population or as a percentage. We calculated PKU prevalence for all considered studies; however, only national programs yielded solid estimates. To report a reliable comparison, we converted PKU prevalence proportions, which in different studies were reported in various units, by percentage. Due to our calculation, Thailand presented the least percentage of PKU prevalence (0.00044%) and Italy faced the most prevalence of PKU, i.e. approximately 0.02736%. Moreover, the prevalence of PKU among patients with ID in Iran was reported by 2.1% in 2009.
A summary of the whole prevalence and incidence of PKU in numerous countries is presented in Tables 1, 2, 3 & 4. Most of the cases included in this study are not only cPKU, but also Hyperphenylalaninemia (HPA) not requiring treatment. Data related to all PKU variants frequency are listed in Tables 1, 2, 3 & 4; however, the relevant frequency of different PKU variants was not calculated in some of the references. Thus, we assumed the PKU frequency in these publications was related to all PKU phenotypes. In the case of the data availability related to the percentage of BH4 responsiveness among the different HPA and PKU variants in the eligible publications that were investigated for this study, this percentage is reported in Tables 1, 2, 3 & 4, as well. Extensive studies are available on the prevalence of PKU in Iran. The incidence of PKU in Iran in 1982 was estimated as 0.011% (23); however, this rate had raised to 0.02757% in 2002 (24), and it had later reduced to 0.02% of live births in 2015. Among these cases, 18.8%, 26.4%, 34%, and 20.8%, respectively presented mHPA (360-600 μmol/L blood Phe), mild PKU (mPKU; 600-900 μmol/L blood Phe), modPKU (900-1200 μmol/L blood Phe), and cPKU (blood Phe >1200 μmol/L) (2). Using HPLC in Fars Province, the south of Iran, the incidence of PKU from 2004 to 2007 was reported to be 0.01597% (25); however, this proportion reduced to 0.01169% from 2007 to -2008 by the screening of a total of 76966 newborns (62.5% mHPA &d 37.5% PKU) (26).
In a descriptive retrospective study in Mazandaran Province, northern Iran, 0.00662% of newborns were diagnosed with PKU from 2007 to 2015 using ELISA and HPLC as the confirmation methods. HPA phenotypes in Mazandaran was reported as 22.2% for cPKU, 33.3% for mPKU, and 44.5% for HPA (13). Another study in patients with ID (except Down syndrome) in Tehran and 31 other cities of Iran exhibited the %2.1 prevalence of cPKU and %0.44 of mHPA cases (27). These studies revealed that despite the implementation of the Newborn Screening (NBS) programs for an Inborn Error of Metabolism (IEM) in most cities of Iran, the prevalence of PKU remained significant.
The frequency of the disease in Asian countries
According to our findings, based on the Ministry of Health, PKU was highly prevalent in Iran (0.02%), particularly in Fars Province with a prevalence of 0.01597% from 2004 to 2007 (25). This rate was respectively higher than Turkey, UAE, Lebanon, Iraq, China, Saudi Arabia, Mazandaran (Iran), India, Bahrain, Taiwan, Singapore, Japan, Korea, and Thailand. Nevertheless, PKU occurrence experienced a reduction to 0.01169% in Fras Province in the following years (2007-2008) (26), i.e. less than PKU incidence in Iraq, Lebanon, UAE, and Turkey. Although in Lebanon from 1998 to 2007, only 18 cases of PKU variants were detected by screening 143000 newborns (0.01258%); after introducing expanded NBS by MS/MS in 2007, the incidence of PKU equaled 0.02613%. This rate reduced to 0.01984% of newborns by 2013 (21, 28); the PKU disorders were of the highest prevalence in this country among the other Asian countries. After Lebanon and Iran, the highest PKU prevalence belonged to Turkey with 0.01538% of newborns in 2007 (29). In the UAE however, the incidence of PKU from 1998 to 2000 was nearly close to Thailand, which had raised from 0.00498% (30) to 0.00687% (89% cPKU & 1.75% PKU) in 1995-2011 (31). PKU prevalence reached 0.01323 % (only cPKU) in 2011-2014; UAE was ranked fourth for having a high prevalence of PKU in the investigated Asian countries (Table 1) (32). Besides, 0.01211% of newborns presented PKU in Sulaimani City in Iraq. Accordingly, Hamawandi et al. claimed that using Iranian kit materials and lab equipment, as well as the similar ethnicity of Fars and Kurdish population might be the possible explanations for this similarity (33).
The rate of PKU incidence in China and Saudi Arabia were more than in Mazandaran Province in Iran and measured as 0.00862% (16) and 0.00702% (14), respectively. In some countries, we only could find outdated data, e.g., the latest PKU incidence report in India was published in 2004 (0.00546%) (34); however, this rate in Southern India was reported as 34.5% of IEM in 2007-2009 (35). The United Kingdom of Bahrain reported the PKU incidence of 0.0045% (36). In Taiwan, from 2001 to 2014, 0.00167% of Taiwanese live births were detected with PKU (37). An 8-year study in Singapore revealed an incidence of 0.00112% for PKU (38). This rate was 0.0008% in Nagasaki, Japan, in 2001 (39). The reason for the low incidence of PKU in Japan was claimed to be because of genetic drift in the founder population of Japanese islands (40). In South Korea, NBS on 3.44 million in 2000-2015 indicated 0.00072% of newborns with PKU (41). Among Asian countries, Thailand possessed the least prevalence of PKU (0.00044%) (15) after Korea. We can suggest the high rate of the heterogeneity of PAH locus (42, 43) and consanguineous marriages among the Iranian and Turkish population (10, 27, 44) as remarkable causes of the high occurrence of PKU; thus, these countries are of high rank in PKU frequency in Asia.
The frequency of the disease in European countries
In Europe, apart from Malta and Finland (because of their known low PKU incidence), all countries use a national program for PKU; although in some countries, these programs fail to have 100% coverage. Italy (0.02736%) (18), and Germany (0.02%) (41) presented the highest frequency of PKU among European countries. Slovak Republic (Bratislava, Banska Bystrica, & Kosice centers) in 2012 and Ireland in 2004 with 0.01694% PKU incidence (45, 46) and 0.01612% PKU prevalence (18) detected among newborns, respectively possessed the highest PKU frequency after Germany and Italy. The incidence of PKU had an estimated 0.01492% of newborns in Estonia from 1974 to 2016 (47), i.e. almost close to Slovenia (0.01477%, cPKU=0.0098%) (46) and PKU prevalence in Latvia (0.01474%) (18). However, the ethnic origin of the reported subjects in Estonia was 26% of Slavic (Russian), 7% of mixed-origin, and the rest were Estonian. Furthermore, as per the State Expert Center MoH, PKU prevalence was reported to be 0.01428% in Ukrainians (48). Moreover, PKU prevalence was documented as 0.01318% for Switzerland (18). This rate is almost close to PKU frequency in Russia (0.01296%), Scotland (0.01281%), Poland (0.01239%), and Croatia (0.01208%; 10% of the patients were classified as mHPA, 23% were assigned to mPKU, and 67% to cPKU) (18, 49).
In other European countries, PKU frequency was reported as 0.01168% in France (50), 0.01% in UK (51), 0.00833% in Austria (52), 0.00872% in Norway (18), 0.00822% in 2010 in Portugal (53), 0.00808% in Spain (HPA=0.016%) (54), 0.00788% in Sweden (18), 0.00788% in Hungary, 0.0077% in Netherlands (18), and 0.00744 in Denmark (18). Although the official incidence of PKU in Romania remains unpublished, it has been reported by Bucharest National Authority for neonatal screening diagnostic confirmation and specific treatment in PKU and congenital hypothyroidism as 0.01% of live births in 2013 (55). PKU incidence in some other European countries is summarized in Table 2. Among European countries, Finland experienced the lowest PKU frequency by an incidence of 0.001-0.0005; however, this is the latest incidence we could find in our database, i.e. related to 2005 (56). Due to our investigation, the highest and lowest rate of PKU distribution belonged to Italy (0.027%) and Finland (0.0007%), respectively. Notably, the incidence of PKU in Germany was respectively 25- and 45-fold higher than that in Japan and Thailand; however, it was equal to the occurrence of this disorder in Iran.
The frequency of the disease in African countries
The prevalence of PKU in Egypt was evaluated as 0.01978% of infants in 2008 (57), by 2013 this rate reduced to 0.00909%. In 2012, PKU was the most common aminoacidopathies disorder among Tunisians as another African country (58). Khemir et al. estimated the incidence of PKU in Tunisia as 0.0131% from 1988 to 2008 (19). Insufficient diagnostic capabilities in Africa might obstruct revealing the actual rate of PKU frequency; in general, the occurrence of PKU has been reported as low in Africa (59). In this regard, the high frequency of PKU in the north of Africa might be owed to the high level of consanguineous marriage (Table 3).
The frequency of the disease in American countries
In 2005, Buenos Aires in Argentina (0.03333%) (60) possessed the highest, and Cuba (0.00191) (61) had the lowest rate of PKU incidence among Latin American countries. Based on the calculated occurrence of IEM in the U.S.A through NBS by the National Newborn Screening Information System (NNSIS), the US witnessed a decrease in PKU incidence from 0.00524% in 1991-2000 to 0.00433% in 2001-2010 (20). By 2014, 10000 individuals in the US were diagnosed with PKU. In 2001-2010, Hawaii was the US state with the least prevalence of PKU (0.00054%) and Wyoming presented the most PKU prevalence (0.016%) (20). In northern America, the PKU incidence rate was reported as 0.00625% (62). Moreover, PKU occurs in 0.01% of Caucasians (9) and 0.00191% of African Americans in the US (63) (Table 4).
Certain restrictions have affected this review. First, the main search was limited to the publications in English. A major disadvantage of this review concerned its design, in particular for prevalence/incidence studies in which data from most of the reports are based on the collection of the retrospective data either from registries or medical records. Such routine data possess some pitfalls, such as imprecision or deficiency. Another possible limitation included the small sample sizes. Other limitations involved the method for calculating PKU prevalence/incidence in the reports. Some reports applied the divisor as several whole live births and not the actual number of screened cases; however, others performed PKU prevalence/incidence evaluation using a total number of abnormal cases instead of total screened subjects. However, in the case of these reports using self-calculations, we tried to provide more precise data of PKU prevalence/incidence.
4. Conclusion
Advances in science and technology drive the evolution of methods, which allow the accurate measurements of acylcarnitines, amino acids, and other critical metabolites in the diagnosis of metabolic disorders, like MS/MS (64). Due to its sensitivity, selectivity, the ability to screen several diseases simultaneously, and short-term analysis, MS/MS has been widely used for NBS (65). Moreover, the widespread use of MS/MS can significantly be contributed to the estimating of the frequency of incidence of each IEM in our population; reports from countries that have incorporated NBS indicated that the occurrence of some disorders is higher than expected (38, 66). Overall, the prevalence of PKU worldwide in 2015 was claimed to be 0.00641% of live births (67). This review aimed to report the PKU prevalence in different countries to raise awareness and leads to establishing an efficient NBS program. Surprisingly, the highest and lowest relevant rates belonged to Asia and a European country, respectively. Therefore, with a slight difference between Iran, Germany, and Italy, the highest rate of incidence of PKU belonged to Italy with approximately 0.02736% of total live newborns. In contrast, the lowest rate of PKU distribution in Asia belonged to Thailand (about 0.0004%), and in Europe, it concerned Finland. Although we could not conclude the prevalence of PKU in Africa just based on two studies, this prevalence in Tunisia was about the same as some European and Asian countries. In conclusion, in the investigated studies, PKU had covered more than 50% of IEMs in Germany and Italy (68, 69), while this rate for Asian countries (Oman, Japan, Bahrain, India, China) ranged between 6% and 20% of IEMs (data not presented) (70). These findings indicated that the incidence rates of amino acids differ between European and Asian populations.
Our research demonstrated the necessity of conducting further investigations on PKU prevalence, especially in Iran and other countries in the Middle East. Thus, the priority is to assist the countries where the basic screening programs encompass less than 100% coverage. Additionally, continuous monitoring of the NBS program could help to decrease the variation in design and methodology using the available knowledge and expertise in the literature. The considerable difference in recalling the PKU incidence rates represents one important and obvious area for the refinement of NBS program performances, globally. Since treatment depends on the early diagnosis of the disorder, the implementation of neonatal screening for PKU is crucial. Moreover, the estimation of the exact prevalence of the disorder is required to implement effective policies and strategies for controlling the disease occurrence.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors equally contributed to preparing this article.
Conflicts of interest
The authors declared no conflicts of interest.
References
- Karam PE, Alhamra RS, Nemer G, Usta J. Spectrum of mutations in Lebanese patients with phenylalanine hydroxylase deficiency. Gene. 2013; 515(1):117-22. [DOI:10.1016/j.gene.2012.11.018] [PMID]
- Setoodeh A, Yarali B, Rabbani A, Khatami S, Shams S. Tetrahydrobiopterin responsiveness in a series of 53 cases of phenylketonuria and hyperphenylalaninemia in Iran. Molecular Genetics and Metabolism Reports. 2015; 2:77-9. [DOI:10.1016/j.ymgmr.2015.01.001] [PMID] [PMCID]
- van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet Journal of Rare Diseases. 2017; 12(1):162. [DOI:10.1186/s13023-017-0685-2] [PMID] [PMCID]
- Campistol J, Gassió R, Artuch R, Vilaseca MA. Neurocognitive function in mild hyperphenylalaninemia. Developmental Medicine and Child Neurology. 2011; 53(5):405-8. [DOI:10.1111/j.1469-8749.2010.03869.x] [PMID]
- van Spronsen FJ, Rijn M, Dorgelo B, Hoeksma M, Bosch AM, Mulder MF, et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. Journal of Inherited Metabolic Disease. 2009; 32(1):27-31. [DOI:10.1007/s10545-008-0937-3] [PMID]
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Molecular Genetics and Metabolism. 2011; 104(suppl.):S2-9. [DOI:10.1016/j.ymgme.2011.08.017] [PMID]
- Blau N, Van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010; 376(9750):1417-27. [DOI:10.1016/S0140-6736(10)60961-0]
- Pourfarzam M, Zadhoush F. Newborn Screening for inherited metabolic disorders: News and views. Journal of Research in Medical Sciences. 2013; 18(9):801-8. [PMCID]
- Schwoerer JS, Drilias N, Kuhl A, Mochal S, Baker M. Genotypes of patients with phenylalanine hydroxylase deficiency in the Wisconsin Amish. Molecular Genetics and Metabolism Reports. 2018; 15:75-7. [DOI:10.1016/j.ymgmr.2018.02.005] [PMID] [PMCID]
- Mokhtari R, Bagga A. Consanguinity, genetic disorders and malformations in the Iranian population. Acta Biologica Szegediensis. 2003; 47(1-4):47-50. https://www2.sci.u-szeged.hu/ABS/2003/ActaHP/4747.pdf
- Soltani Z, Karami F, Yassaee V, Hashemi Gorji F, Talebzadeh M, Miryounesi M. First case report of EX3del4765 mutation in PAH gene in Asian population. Iranian Red Crescent Medical Journal. 2016; 18(2):e21633. [DOI:10.5812/ircmj.21633] [PMID] [PMCID]
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Medicine. 2009; 6(7):e1000097. [DOI:10.1371/journal.pmed.1000097] [PMID] [PMCID]
- Abbaskhanian A, Zamanfar D, Afshar P, Asadpoor E, Rouhanizadeh H, Jafarnia A, et al. Incidence of Neonatal Hyperphenylalaninemia Based on High-performance Liquid Chromatography Confirmatory Technique in Mazandaran Province, Northern Iran (2007-2015). International Journal of Preventive Medicine. 2017; 8:93. [DOI:10.4103/ijpvm.IJPVM_24_17] [PMID] [PMCID]
- Alfadhel M, Al Othaim A, Al Saif S, Al Mutairi F, Alsayed M, Rahbeeni Z, et al. Expanded Newborn Screening Program in Saudi Arabia: Incidence of screened disorders. Journal of Paediatrics and Child Health. 2017; 53(6):585-91. [DOI:10.1111/jpc.13469] [PMID]
- Banpavichit A, Wiwanitkit V, Sutivijit Y. Prevalence of neonatal hypothyroidism and phenylketonuria in Southern Thailand: A 10-year report. Indian Journal of Endocrinology and Metabolism. 2011; 15(2):115-7. [DOI:10.4103/2230-8210.81941] [PMID] [PMCID]
- Gu X, Wang Z, Ye J, Han L, Qiu W. Newborn screening in China: Phenylketonuria, congenital hypothyroidism and expanded screening. Annals Academy of Medicine. 2008; 37(12):107-10. https://www.annals.edu.sg/pdf/37VolNo12SupplDec2008/V37N12(Suppl)p107.pdf
- Zerjav Tansek M, Groselj U, Angelkova N, Anton D, Baric I, Djordjevic M, et al. Phenylketonuria screening and management in southeastern Europe: Survey results from 11 countries. Orphanet Journal of Rare Diseases. 2015; 10(1):68. [DOI:10.1186/s13023-015-0283-0] [PMID] [PMCID]
- Loeber JG. Neonatal screening in Europe: The situation in 2004. Journal of Inherited Metabolic Disease. 2007; 30(4):430-8. [DOI:10.1007/s10545-007-0644-5] [PMID]
- Khemir S, El Asmi M, Sanhaji H, Feki M, Jemaa R, Tebib N, et al. Phenylketonuria is still a major cause of mental retardation in Tunisia despite the possibility of treatment. Clinical Neurology and Neurosurgery. 2011; 113(9):727-30. [DOI:10.1016/j.clineuro.2011.07.016] [PMID]
- Therrell BL, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Molecular Genetics and Metabolism. 2014; 113(1):14-26. [DOI:10.1016/j.ymgme.2014.07.009] [PMID] [PMCID]
- Karam PE, Habbal M, Mikati MA, Zaatari GE, Cortas NK, Daher RT. Diagnostic challenges of aminoacidopathies and organic acidemias in a developing country: A twelve-year experience. Clinical Biochemistry. 2020; 46(18):1787-92. [DOI:10.1016/j.clinbiochem.2013.08.009] [PMID]
- Selim LA, Hassan SA-HH, Salem F, Orabi A, Hassan FA, El-Mougy F, et al. Selective screening for inborn errors of metabolism by tandem mass spectrometry in Egyptian children: A 5-year report. Clinical Biochemistry. 2014; 47(9):823-8. [DOI:10.1016/j.clinbiochem.2014.04.002] [PMID]
- Farhud DD, Kabiri M. Incidence of phenylketonuria (PKU) in Iran. Indian Journal of Pediatrics. 2010; 49(400):685-8. [DOI:10.1007/BF02752654] [PMID]
- Koochmeshgi J, Bagheri A, Hosseini-Mazinani SM. Incidence of phenylketonuria in Iran estimated from consanguineous marriages. Journal of Inherited Metabolic Disease. 2002; 25:80-1. [DOI:10.1023/A:1015154321142] [PMID]
- Habib A, Fallahzadeh MH, Kazeroni HR, Ganjkarimi AH. Incidence of phenylketonuria in Southern Iran. Iranian Journal of Medical Sciences (IJMS). 2010; 35(2):137-9. https://ijms.sums.ac.ir/article_39771.html
- Karamifar H, Ordoei M, Karamizadeh Z, Amirhakimi G. Incidence of neonatal hyperphenylalaninemia in Fars Province, south Iran. Iranian Journal of Pediatrics. 2010; 20(2):216-20. [PMID] [PMCID]
- Ghiasvand NM, Aledavood AR, Ghiasvand R, Seyedin Borojeny F, Aledavood AR, Seyed S, et al. Prevalence of classical phenylketonuria in mentally retarded individuals in Iran. Journal of Inherited Metabolic Disease. 2009; 32(suppl 1):283-7. [DOI:10.1007/s10545-009-1222-9] [PMID]
- Khneisser I, Adib S, Assaad S, Megarbane A, Karam P. Cost-benefit analysis: Newborn screening for inborn errors of metabolism in Lebanon. Journal of Medical Screening. 2020; 22(4):182-6. [DOI:10.1177/0969141315590675] [PMID]
- Dobrowolski SF, Heintz C, Miller T, Ellingson CC, Ellingson CC, Özer II, et al. Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population. Molecular Genetics and Metabolism. 2011; 102(2):116-21. [DOI:10.1016/j.ymgme.2010.11.158] [PMID]
- Al-Hosani H, Salah M, Saade D, Osman H, Al-Zahid J. United Arab Emirates National Newborn Screening Programme: An evaluation 1998-2000. Eastern Mediterranean Health Journal (EMHJ). 2003; 9(3):324-32. https://apps.who.int/iris/handle/10665/119281
- Al Hosani H, Salah M, Osman HM, Farag HM, El Assiouty L, Saade D, et al. Expanding the comprehensive national neonatal screening programme in the United Arab Emirates from 1995 to 2011. Eastern Mediterranean Health Journal (EMHJ). 2014; 20(1):17-23. [DOI:10.26719/2014.20.1.17]
- Al-Jasmi FA, Al-Shamsi A, Hertecant JL, Al-Hamad SM, Souid AK. Inborn errors of metabolism in the United Arab Emirates: Disorders detected by newborn screening (2011-2014). JIMD Reports. 2016; 28:127-35. [DOI:10.1007/8904_2015_512] [PMID] [PMCID]
- Hamawandi AMH, Ahmed Rashid J, Heersh Raof Saeed HMH, Hawrami OM. Annual incidence of phenylketonuria in Sulaimani City. Merit Research Journals. 2015; 3(9):427-31. https://www.researchgate.net/publication/282353380_Annual_incidence_of_phenylketonuria_in_Sulaimani_City
- Rama Devi AR, Naushad SM, Division D. Newborn screening in India. Indian Journal of Pediatrics. 2004; 71(2):157-60. [DOI:10.1007/BF02723099] [PMID]
- Nagaraja D, Mamatha SN, De T, Christopher R. Screening for inborn errors of metabolism using automated electrospray tandem mass spectrometry: Study in high-risk Indian population. Clinical Biochemistry. 2010; 43(6):581-8. [DOI:10.1016/j.clinbiochem.2009.12.009] [PMID]
- Golbahar J, Al-Jishi EA, Altayab DD, Carreon E, Bakhiet M, Alkhayyat H. Selective newborn screening of inborn errors of amino acids, organic acids and fatty acids metabolism in the Kingdom of Bahrain. Molecular Genetics and Metabolism. 2013; 110(1-2):98-101. [DOI:10.1016/j.ymgme.2013.07.006] [PMID]
- Niu DMM, Chien YHH, Chiang CCC, Ho HCC, Hwu W-LL, Kao S-MM, et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. Journal of Inherited Metabolic Disease. 2010; 33(suppl. 2):295-305. [DOI:10.1007/s10545-010-9129-z] [PMID]
- Lim JS, Tan ETHHSTH, John CM, Poh S, Yeo SJ, Ang JSMM, et al. Inborn error of metabolism (IEM) screening in Singapore by electrospray ionization-tandem mass spectrometry (ESI/MS/MS): An 8year journey from pilot to current program. Molecular Genetics and Metabolism. 2014; 113(1):53-61. [DOI:10.1016/j.ymgme.2014.07.018] [PMID]
- El-Metwally A, Yousef Al-Ahaidib L, Ayman Sunqurah A, Al-Surimi K, Househ M, Alshehri A, et al. The prevalence of phenylketonuria in Arab countries, Turkey, and Iran: A Systematic Review. BioMed Research International. 2018; 2018:7697210. [DOI:10.1155/2018/7697210] [PMID] [PMCID]
- Okano Y, Hase Y, Lee D-H, Furuyama J-I, Shintaku H, Oura T, et al. Frequency and distribution of phenylketonuric mutations in orientals. Human Mutation. 1992; 1(3):216-20. [DOI:10.1002/humu.1380010307] [PMID]
- Shibata N, Hasegawa Y, Yamada K, Kobayashi H, Purevsuren J, Yang Y, et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective screening vs expanded newborn screening. Molecular Genetics and Metabolism. 2018; 16:5-10. [DOI:10.1016/j.ymgmr.2018.05.003] [PMID] [PMCID]
- Biglari A, Saffari F, Rashvand Z, Alizadeh S, Najafipour R, Sahmani M. Mutations of the phenylalanine hydroxylase gene in Iranian patients with phenylketonuria. SpringerPlus. 2015; 4(1):122-30. [DOI:10.1186/s40064-015-1309-8] [PMID] [PMCID]
- Zamanfar D, Jalali H, Mahdavi MR, Maadanisani M, Zaeri H, Asadpoor E. Investigation of five common mutations on phenylalanine hydroxylase gene of phenylketonuria patients from two provinces in north of Iran. International Journal of Preventive Medicine. 2017; 8:89.[DOI: 10.4103/ijpvm.IJPVM_378_16] [PMID] [PMCID]
- Morovatdar N, Badiee Aval S, Hosseini Yazdi SMR, Norouzi F, Mina T. The epidemiological and clinical study of Phenylketonuria (PKU) patients in Khorasan, north-eastern Iran. Iranian Journal of Neonatology. 2015; 6(1):18-22. https://www.researchgate.net/publication/275355224_The_Epidemiological_and_Clinical_Study_of_Phenylketonuria_PKU_Patients_in_Khorasan_North-eastern_Iran
- Polak E, Ficek A, Radvanszky J, Soltysova A, Urge O, Cmelova E, et al. Phenylalanine hydroxylase deficiency in the Slovak population: Genotype-phenotype correlations and genotype-based predictions of BH4-responsiveness. Gene. 2013; 526(2):347-55. [DOI:10.1016/j.gene.2013.05.057] [PMID]
- Šmon A, Grošelj, UrhTanšek, Mojca ŽerjavBiček, AjdaOblak, AdrijanaZupančič et al. Newborn screening in Slovenia. Zdr Varst. 2015; 54(2):86-90. [DOI:10.1515/sjph-2015-0013] [PMID] [PMCID]
- Lillevali H, Reinson K, Muru K, Simenson K, Murumets U, Mols T, et al. Hyperphenylalaninaemias in Estonia: Genotype-phenotype correlation and comparative overview of the patient cohort before and after nation-wide neonatal screening. JIMD Reports. 2018; 40:39-45. [DOI:10.1007/8904_2017_61] [PMID] [PMCID]
- Pampukha V, Nechyporenko M, Livshyts L. Analysis of EX5del4232ins268 and EX5del955 PAH gene mutations in Ukrainian patients with phenylketonuria. Genes Dis. 2017; 4(2):108-10. [DOI:10.1016/j.gendis.2016.11.004] [PMID] [PMCID]
- Karačić I, Meili D, Sarnavka V, Heintz C, Thöny B, Ramadža DP, et al. Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian Patients with Phenylalanine Hydroxylase (PAH) deficiency. Molecular Genetics and Metabolism. 2009; 97(3):165-71. [DOI:10.1016/j.ymgme.2009.03.009] [PMID]
- Wiedemann A, Leheup B, Battaglia-Hsu SF, Jonveaux P, Jeannesson E, Feillet F. Undiagnosed phenylketonuria in parents of phenylketonuric patients, is it worthwhile to be checked? Molecular Genetics and Metabolism. 2013; 110:62-5. [DOI:10.1016/j.ymgme.2013.08.014] [PMID]
- MacDonald A, Smith TA, de Silva S, Alam V, van Loon JMT. The personal burden for caregivers of children with phenylketonuria: A cross-sectional study investigating time burden and costs in the UK. Mol Genet Metab Reports. 2016; 9:1-5. [DOI:10.1016/j.ymgmr.2016.08.008] [PMID] [PMCID]
- Kasper DC, Ratschmann R, Metz TF, Mechtler TP, Möslinger D, Konstantopoulou V, et al. The National Austrian newborn screening program: Eight years experience with mass spectrometry. Past, present, and future goals. Wien Klin Wochenschr. 2010; 122(21-22):607-13. [DOI:10.1007/s00508-010-1457-3] [PMID]
- Vilarinho L, Rocha H, Sousa C, Marcão A, Fonseca H, Bogas M, et. al. Four years of expanded newborn screening in Portugal with with tandem mass spectrometry. Journal of Inherited Metabolic Disease. 2010; 33(suppl 3):S133-8. [DOI:10.1007/s10545-010-9048-z] [PMID]
- Couce ML, Castiñeiras DE, Bóveda MD, Baña A, Cocho JA, Iglesias AJ, et al. Evaluation and long-term follow-up of infants with inborn errors of metabolism identified in an expanded screening programme. Molecular Genetics and Metabolism. 2011; 104(4):470-5. [DOI:10.1016/j.ymgme.2011.09.021] [PMID]
- Gemperle-Britschgi C, Iorgulescu D, Mager MA, Anton-Paduraru D, Vulturar R, Thöny B. A novel common large genomic deletion and two new missense mutations identified in the Romanian phenylketonuria population. Gene. 2016; 576(1):182-8. [DOI:10.1016/j.gene.2015.10.020] [PMID]
- Autti-Rämö I, Måkelå M, Sintonen H, Koskinen H, Laajalahti L, Halila R, et al. Expanding screening for rare metabolic disease in the newborn: An analysis of costs, effect and ethical consequences for decision-making in Finland. Acta Paediatr. 2005; 94(8):1126-36. [DOI:10.1111/j.1651-2227.2005.tb02056.x] [PMID]
- Hassan FA, El-mougy F, Sharaf SA, Mandour I, Morgan MF, Selim LA, et al. Inborn errors of metabolism detectable by tandem mass spectrometry in Egypt: The first newborn screening pilot study. Journal of Medical Screening. 2016; 0(0):1-6. [DOI:10.1177/0969141315618229] [PMID]
- Hadj-Taieb S, Nasrallah F, Hammami MB, Elasmi M, Sanhaji H, Moncef F, et al. Aminoacidopathies and organic acidurias in Tunisia: A retrospective survey over 23 years. La Tunisie Médicale. 2012; 90(3):258-61. https://www.researchgate.net/publication/223962760_Aminoacidopathies_and_organic_acidurias_in_Tunisia_A_retrospective_survey_over_23_years
- National Institutes of Health (US). Phenylketonuria (PKU): Screening and management. NIH Consens Statement. 2003; 17(3). [PMID]
- Penchaszadeh VB. Genetic testing and services in Argentina. Journal of Community Genetics. 2013; 4(3):343-54. [DOI:10.1007/s12687-012-0093-1] [PMID] [PMCID]
- González EC, Frómeta A, del Río L, Castells E, Robaina MS, García SM, et al. Cuban neonatal screening of phenylketonuria using an ultramicro-fluorometric test. Clinica Chimica Acta. 2009; 402(1-2):129-32. [DOI:10.1016/j.cca.2008.12.039] [PMID]
- Hamman KJ, Winn SR, Harding CO. Hepatocytes from wild-type or heterozygous donors are equally effective in achieving successful therapeutic liver repopulation in murine phenylketonuria (PKU). Molecular Genetics and Metabolism. 2011; 104(3):235-40. [DOI:10.1016/j.ymgme.2011.07.027] [PMID] [PMCID]
- Hofman KJ, Steel G, Kazazian HH, Valle D. Phenylketonuria in U.S. blacks: Molecular analysis of the phenylalanine hydroxylase gene. American Journal of Human Genetics. 1991; 48(4):791-8. https://jhu.pure.elsevier.com/en/publications/phenylketonuria-in-us-blacks-molecular-analysis-of-the-phenylalan-5
- Levy HL. Newborn screening by tandem mass spectrometry: A new era. Clinical Chemistry. 1998; 44(12):2401-2. [DOI:10.1093/clinchem/44.12.2401] [PMID]
- Chace DH. Mass spectrometry in newborn and metabolic screening: Historical perspective and future directions. Journal of Mass Spectrometry (JMS). 2009; 44(2):163-70. [DOI:10.1002/jms.1528] [PMID]
- Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. The New England Journal of Medicine. 2003; 348(23):2304-12. [DOI:10.1056/NEJMoa025225] [PMID]
- Verma IC, Puri RD. Global burden of genetic disease and the role of genetic screening. Seminars in Fetal and Neonatal Medicine. 2015; 20(5):354-63. [DOI:10.1016/j.siny.2015.07.002] [PMID]
- Lindner M, Gramer G, Haege G, Fang-Hoffmann J, Schwab KO, Tacke U, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases: Report of 10 years from South-West Germany. Orphanet Journal of Rare Diseases. 2011; 6(1). [DOI:10.1186/1750-1172-6-44] [PMID] [PMCID]
- La Marca G, Malvagia S, Casetta B, Pasquini E, Donati MA, Zammarchi E. Progress in expanded newborn screening for metabolic conditions by LC-MS/MS in Tuscany: Update on methods to reduce false tests. Journal of Inherited Metabolic Disease. 2008; 31(suppl. 2):395-404. [DOI:10.1007/s10545-008-0965-z] [PMID]
- Al Riyami S, Al Maney M, Joshi SN, Bayoumi R. Detection of inborn errors of metabolism using tandem mass spectrometry among high-risk Omani patients. Oman Medical Journal. 2012; 27(6):482-5. [DOI:10.5001/omj.2012.115] [PMID] [PMCID]
- Georgiou T, Ho G, Vogazianos M, Dionysiou M, Nicolaou A, Chappa G, et al. The spectrum of mutations identified in Cypriot patients with phenylalanine hydroxylase deficiency detected through neonatal screening. Clinical Biochemistry. 2012; 45(7-8):588-92. [DOI:10.1016/j.clinbiochem.2012.01.026] [PMID]
- Cornejo V, Raimann E, Cabello JF, Valiente A, Becerra C, Opazo M, et al. Past, present and future of newborn screening in Chile. Journal of Inherited Metabolic Disease. 2010; 33(suppl. 3):301-6. [DOI:10.1007/s10545-010-9165-8] [PMID]
Type of Study: Systematic Review |
Subject:
Pediatric Endocrinology
Received: 2020/07/27 | Accepted: 2020/10/11 | Published: 2021/04/1
Received: 2020/07/27 | Accepted: 2020/10/11 | Published: 2021/04/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
