
Sun, Jun 14, 2026


Volume 12, Issue 1 (1-2024)
J. Pediatr. Rev 2024, 12(1): 87-94 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Mohajeri-Tehrani M, Arab F, Salemkar S, Darvishian N, Mohseni F, Larijani B et al . Investigating β Thalassemia Patients and Their Growth: A Brief Review and Our Clinical Experience. J. Pediatr. Rev 2024; 12 (1) :87-94
URL: http://jpr.mazums.ac.ir/article-1-503-en.html
URL: http://jpr.mazums.ac.ir/article-1-503-en.html
Mohammad-Reza Mohajeri-Tehrani1 

 , Faezeh Arab1 , Sedigheh Salemkar1 , Najmeh Darvishian1 , Fariba Mohseni1 , Bagher Larijani1 , Zohreh Hamidi *2
, Faezeh Arab1 , Sedigheh Salemkar1 , Najmeh Darvishian1 , Fariba Mohseni1 , Bagher Larijani1 , Zohreh Hamidi *2
, Faezeh Arab1 , Sedigheh Salemkar1 , Najmeh Darvishian1 , Fariba Mohseni1 , Bagher Larijani1 , Zohreh Hamidi *2
1- Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
2- Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran. ,zohreh.hamidi@gmail.com
2- Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran. ,
Full-Text [PDF 410 kb]
(1919 Downloads)
| Abstract (HTML) (3141 Views)
Full-Text: (1235 Views)
Introduction
The first comprehensive and global study on human growth diversity is performed by Eveleth and Tanner. According to the limitations of that time, their research was a descriptive study. They did not agree with a previous study that the growth of all populations can be based on one universal reference. They suggested that there are no guarantees that all children have the same growth potential. Based on their study, the growth pattern of children of the same age is different due to genetic variation [1].
Their work explained growth patterns in normal children but growth and growth deceleration problems in chronic disease is another subject. Thalassemia major is a chronic disease and it is not an exception in this subject. The prevalence of short stature is reported around 31% to 55% in these patients [2-7].
β-thalassemia major is one of the most severe forms of these disorders. The inability of the body to produce β chain of hemoglobin leads to β thalassemia diseases. Patients need frequent blood transfusions during their lifetime [8] and blood transfusions in long-term periods cause iron overload.
Iron overload and secondary hemochromatosis that happen following frequent blood transfusion lead to the production of free radicals and oxidative destruction with numerous complications, including heart failure, liver cirrhosis, and endocrine disorders such as hypogonadism, hypothyroidism, diabetes, calcium homeostasis disturbances and bone disease, fatigue, joint pain and darkening of the skin occur.
The incidence of hypothyroidism, diabetes, impaired fasting glucose, and hypogonadism among β-thalassemia patients were reported from 6.6% to 16%, 5% to 8%, 26% to 27%, and 16% to 44%, respectively [2, 5, 9-11]. In addition, the rate of low bone mass density and fracture was 10% to 51% and 30% to 50% among patients [2, 5, 12-14].
Endocrine glands are more susceptible to iron overload and even small increases in iron in the early stage of life can cause many disturbances [9]. The structure of the pituitary gland in the first four years of life becomes abnormal and the rate of growth hormone secretion also reduces due to the high amount of iron [15]. The production of insulin-like growth factor 1 (IGF 1), whole growth hormone IGF 1 and insulin-like growth factor-binding protein 3, secondary due to liver hemosiderosis or chronic viral hepatitis may also decrease [16].
Increasing accumulation of iron in the body decreases sex and growth hormone secretion which are the main reasons for short stature in β-thalassemic patients. Other main reasons for short stature in β-thalassemic children are hypothyroidism, decreased bone density (mainly in the spine) and bone deformity [1, 15-17].
Malnutrition is another problem. Apart from low economic status or gastrointestinal disorders, there are also other factors contributing to malnutrition. Hyper-metabolic status in β-thalassemia patients and difficulty in managing a sufficiently balanced diet are important. Reducing the consumption of food containing iron will reduce the intake of adequate calories. The recommendation for tea consumption to reduce iron absorption leads to a reduction in milk consumption, thereby increasing the essential nutrients for normal growth, such as protein, calcium, and zinc decreases [18].
Other contributing factors in growth retardation are pre-transfusion of hemoglobin level [19], lack of folic acid [16], frequent use of chelating drugs [20], emotional factors [8], low socioeconomic level [21], autoimmune factors [22], and genotype of thalassemia [7].
Regarding chelating agents, desferrioxamine is a chelator agent that is intensive and or premature use (for example, before 3 years of age and >50 mg/kg/day), and may have bone complications [23]. Taking these agents in patients with hyper transfusion is necessary because desferrioxamine binds to iron and removes excess iron from the body; however, it may cause metaphyseal long-bone dysplasia, spinal changes, and growth retardation. Desferrioxamine also prevents the synthesis of DNA and cell proliferation, collagen formation, and degradation of copper, zinc or calcium in the bone matrix. The lack of essential micronutrients reduces alkaline phosphatase activity, and decreases spinal height, thereby leading to truncal shortening, even in the presence of normal stature [24]. Body disproportion between the upper and lower body segments (the former is shorter than normal and the latter is normal) is a common feature of short stature in thalassemia patients. Male sex is suggested as an independent risk factor [19]. However, several studies argue otherwise [25-27].
The exact age of growth retardation in β-thalassemia patients does not have the same depth of research for its correlated results. According to Saxena, growth retardation occurs at the age of 8 years [21]. Therefore, most children with thalassemia have normal growth during the childhood period; however, Soliman et al. claimed that the process of growth development declines at the age of 4 [16]. Nokeaingtong et al. reported that in children with thalassemia, the mean height Z-score is reduced by 0.19 SD yearly from 5 to 14 years of age [19]. Another study that was performed in 1973 showed that short stature in boys begins at the age of 8 years and at 11 years old becomes visible. This process in girls begins at 10 years of age and becomes visible at 12 years [21].
Growth development occurs in two stages. The first occurs during the childhood period which depends on growth hormones and the second stage is during the puberty period which depends both on growth and sex hormone secretion [1, 16, 21]. In children with β-thalassemia, growth retardation occurs in both stages of childhood and puberty. However, there is a study which claimed that growth retardation in children with β-thalassemia establishes within three phases. The first phase starts in children with the age of less than 5 years because of ineffective erythropoiesis and anemia. The second phase happens between the ages of 5 to 9 years due to iron overload which affects the pituitary gland and the last phase occurs in the pre-pubertal period [18].
As different cut-off points for growth retardation in thalassemia patients are reported and short stature is more treatable in early periods, it is desirable to find a careful cut-off age for its incidence and help patients before it is too late. In this study, we tried to find such a cut-off point via statistical analysis.
Methods
Study protocol
In this cross-sectional study, data from 803 β-thalassemia major patients were analyzed. Information was extracted from questionnaires from a bone mineral densitometry (BMD) department of a referral center from around the country. The location was the Special Medical Center of Charity Foundation for Special Diseases. The questionnaires were completed during an in-person interview by a BMD operator during 2003-2010. The exclusion criteria were being a current user or chronic user of systemic steroids. Also, patients who reported a major risk factor for osteoporosis were omitted from this study. The inclusion criteria referred to the BMD department to measure height and weight carefully and with one device.
Height and weight were measured by one device as all of the BMD scans were done by one DXA machine (Norland XR-46 densitometer). A medical history was obtained about any drug or disease that positively or negatively affects BMD.
Short stature in children was defined by a height percentile of less than 3%. Hypothyroidism was defined as the current use of levothyroxine. A history of diabetes was based on the suggestion of patients.
Low bone mass was considered as Z scores ≤−2 BMD relative to age and sex-specific norms.
Deformities were reported by the BMD operator. The operator reported it in the spine or femur if deformity remained after a good positioning of patients and did not disappear after careful repositioning.
As diabetes and hypothyroidism (levothyroxine use is regarded as a surrogate of hypothyroidism), are a minor risk factor, patients with a medical history of diabetes and levothyroxine use were not omitted. On the other hand, these two diseases are known disorders by people with medications that increase the accuracy of data received from patients.
The procedures were approved by the Ethics Committee of the Endocrinology and Metabolism Research Institute of Tehran University of Medical Sciences (EMRI of TUM) and performed according to the ethical standards in the 1964 Declaration of Helsinki and its later amendments [28]. The data used for this project were obtained from routine and scientific investigations of thalassemia patients. Moreover, considering the confidentiality of this information, it does not seem to be harmful to the patients. Also, considering that the implementation of this project helps to check the health and help the health of other thalassemia patients, it seems that conducting this study without the consent letter of the patients does not create an ethical problem.
Statistical analysis
SPSS software, version 16 used for statistical analysis. Continuous variables are summarized as Means±SD and ranges. Categorical variables are provided as simple percentages. The independent samples t-test procedure was used for the comparison of means for the two groups of cases. To assess the association between dichotomous variables we used the crosstab. Statistical significance was set at 0.05.
We used an odd ratio (OR) calculation to find the cut-off age for the incidence of short stature. An odds ratio is a relative measure of effect, which allows the comparison of the intervention group of a study relative to the comparison or placebo group. Accordingly, if the outcome is the same in both groups, the ratio will be 1, which implies there is no difference between the two arms of the study. OR calculation usually contains a confidence interval (CI) that if it crosses 1, is non-significant. On the other hand, if the CI does not cross 1, it is significant. This means that one arm of the study (for example, being at an age and higher), significantly (with a 5% CI) changes the risk of an outcome (e.g. diabetes in thalassemic patients). The primary independent parameter (for example, age) must have a significant correlation with the dependent parameter (being diabetic). We used such analysis to find the cut-off age for the incidence of endocrine complications in thalassemia patients. We tried to find that being at what age (and older) significantly raises the risk of any complication that was found significantly related to age (at first, we tested this correlation).
Results
Participants’ characteristics and the prevalence of disorders
The mean age of patients was 20±7 years old and female to male ratio was 420/383. Children (<20 years of age) formed 47% of the participants. The mean ages of males and females were 20.1 and 19.9 years, respectively (no significant difference, P=0.693). Short stature was found in 32% of patients and the youngest short stature patient was 6 years old. Diabetes and hypothyroidism were found in 3.3% and 2% of cases, respectively. The prevalence of low BMD of the neck and spine was almost equal and found in 30% of the participants.
Associations
Significant correlations were found between age, sex, diabetes, and short stature (P≤0.001, 0.042 and <0.001, respectively). Male sex was a risk factor for short stature.
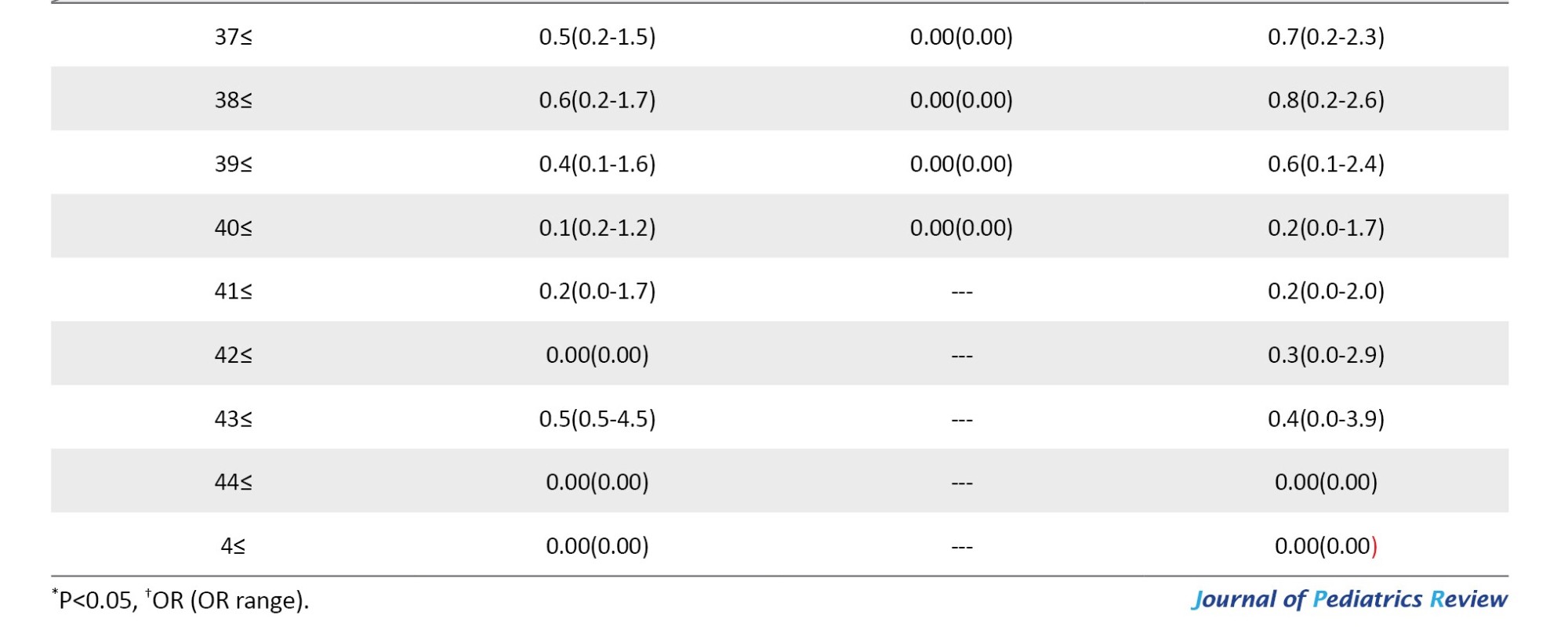
Age has a significant correlation with short stature and it is a continuous parameter. An OR calculation was used to find which age significantly increases the chance of being short and considered it as the cut-off age for short stature. Being 10 and 11 years old and above increased the risk of short stature, 28.5 and 21.4 times, respectively compared to younger patients (both P<0.001). On the other hand, after 7 years of age up to 18 years of age, similar to 10 years of age and 11 years of age, the risk of short stature was increased. Also, being 27 years old and 30 years old and above increases the risk of short stature by 1.4 and 1.7 times, respectively, compared to younger patients (P=0.042 and <0.018, respectively). In Table 1, significant increases in the risk of short stature according to age levels are listed. In this table, we also showed the results for men and women, separately.

Discussion
This study found the prevalence of short stature as high as 32%. Meanwhile, the prevalence of short stature was reported previously at 31% to 55% [2]. Accordingly, the prevalence of short stature in our patients is among the lowest rates. This can be considered the result of the implementation of an active health network service for thalassemic patients in Iran. The thalassemia prevention program was established in 1995. It is extended all over the country (it is based in 64 medical universities and faculties). Almost all thalassemia patients are supported by these centers. It is reported that the mean hemoglobin of patients on the day of transfusion is almost 8.0 g/dL. This can be translated to a relatively good distribution of blood supply for them. Besides, for appropriate management of iron overload, iron chelation agents that are internally manufactured, are free for all thalassemic patients.
In previous studies, 2 and 3 phases of growth retardation were reported in thalassemia patients. Our findings are closer to authors that suggest 3 phases or cut-offs for growth retardation because we found ages of 7 years, 11 years and 18 years as cut-off points. However, according to our findings, growth retardation in thalassemic patients is a continuous phenomenon. Maybe the reason behind this is the incident of deformities due to micro compression fractures of vertebrae, a continuous problem. In this cross-section, we found 4% deformity in the spine 1% deformity in the femur, and 30% of low BMD. However, the results may be different from one ethnicity to another [29].
Male to female difference in short stature prevalence is significant and males have more short stature than females. This is in line with the study by Nokeaingtong et al. They found that children’s height-for-age Z-scores decreased up to the age of 14 (from the age of 5). But at 14 years of age, females showed a growth spurt, whereas growth in male children continued to decline. Maybe females show a better response to hormones or less detrimental reactions to iron overload. Even some investigators suggested they have lesser sensitivity to chronic oxidative stress [19]. But there are studies in disagreement [25-27]. In a study by Altincik al., short stature was present in 42% and in men and women, the prevalence was almost the same [25]. In another population of 40 females and 40 males of β thalassemia patients, the number of short-stature patients was almost the same (33 male, 32 female) [26]. Rathaur et al. found that 14/24 females and 32/46 males were short, but no significant association was found between sex and short stature [27].
Diabetes was found to have a direct correlation with short stature while hypothyroidism did not correlate. We found no scientific reason for these correlations but the late peak of increased risk of short stature at 27 and 30 years of age may be due to an increase in diabetes risk at these ages in thalassemic patients [30].
Conclusion
We suggest that awareness about short stature and monitoring for it in thalassemic patients should be done in their childhood. Given that being 7 years old increases the risk of short stature, we suggest the start of monitoring as early as 5 years of age. We hope that with these studies we improve thalassemic patients’ management while enabling a better quality of life for them.
Study limitations
This study faced some limitations. The lack of information about hypogonadism and delayed puberty in patients was a limitation (data about females is consistent but no question about males’ age of puberty or hypogonadism was asked, so we ignored all data about hypogonadism and delayed puberty). Also, a DXA scan is not the gold standard for diagnosis of spinal or femoral deformity, but if we do not find any deformity in the spinal region in a DXA scan, it is confirmatory. Another limitation of our study was that no data about hormonal lab tests existed in our questionnaire. Although there are some limitations, this study has strong aspects, such as a high number of patients and measurement in a referral center, by one device, and one operator (95% measurement done by one operator). Another novelty of this study is using statistical tools for finding cut-off points of short stature, that were not used or not referred to in previous studies. The statistical test that we used for defining cut-off points in this study is also important, because by using it, we could determine the OR between short stature and age cut-off points that we determined.
Ethical Considerations
Compliance with ethical guidelines
The procedures were approved by the Ethics Committee of the Endocrinology and Metabolism Research Institute of Tehran University of Medical Sciences (EMRI of TUM) (Code: EC-00355) and performed according to the ethical standards in the 1964 Declaration of Helsinki and its later amendments
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Study design: Fariba Mohseni, Mohammad-Reza Mohajeri-Tehrani, Bagher Larijani and Zohreh Hamidi; Study conduct: Zohreh Hamidi; Data collection: Najmeh Darvishian, Faezeh Arab, Sedigheh Salemkar and Zohreh Hamidi; Data analysis: Zohreh Hamidi; Data interpretation: Mohammad Reza Mohajeri-Tehrani, Fariba Mohseni and Zohreh Hamidi; Drafting the manuscript: Zohreh Hamidi; Revising manuscript content: Fariba Mohseni and Mohammad-Reza Mohajeri-Tehrani; Final approval: Mohammad-Reza Mohajeri-Tehrani.
Conflicts of interest
The authors declared no conflict of interest.
References
The first comprehensive and global study on human growth diversity is performed by Eveleth and Tanner. According to the limitations of that time, their research was a descriptive study. They did not agree with a previous study that the growth of all populations can be based on one universal reference. They suggested that there are no guarantees that all children have the same growth potential. Based on their study, the growth pattern of children of the same age is different due to genetic variation [1].
Their work explained growth patterns in normal children but growth and growth deceleration problems in chronic disease is another subject. Thalassemia major is a chronic disease and it is not an exception in this subject. The prevalence of short stature is reported around 31% to 55% in these patients [2-7].
β-thalassemia major is one of the most severe forms of these disorders. The inability of the body to produce β chain of hemoglobin leads to β thalassemia diseases. Patients need frequent blood transfusions during their lifetime [8] and blood transfusions in long-term periods cause iron overload.
Iron overload and secondary hemochromatosis that happen following frequent blood transfusion lead to the production of free radicals and oxidative destruction with numerous complications, including heart failure, liver cirrhosis, and endocrine disorders such as hypogonadism, hypothyroidism, diabetes, calcium homeostasis disturbances and bone disease, fatigue, joint pain and darkening of the skin occur.
The incidence of hypothyroidism, diabetes, impaired fasting glucose, and hypogonadism among β-thalassemia patients were reported from 6.6% to 16%, 5% to 8%, 26% to 27%, and 16% to 44%, respectively [2, 5, 9-11]. In addition, the rate of low bone mass density and fracture was 10% to 51% and 30% to 50% among patients [2, 5, 12-14].
Endocrine glands are more susceptible to iron overload and even small increases in iron in the early stage of life can cause many disturbances [9]. The structure of the pituitary gland in the first four years of life becomes abnormal and the rate of growth hormone secretion also reduces due to the high amount of iron [15]. The production of insulin-like growth factor 1 (IGF 1), whole growth hormone IGF 1 and insulin-like growth factor-binding protein 3, secondary due to liver hemosiderosis or chronic viral hepatitis may also decrease [16].
Increasing accumulation of iron in the body decreases sex and growth hormone secretion which are the main reasons for short stature in β-thalassemic patients. Other main reasons for short stature in β-thalassemic children are hypothyroidism, decreased bone density (mainly in the spine) and bone deformity [1, 15-17].
Malnutrition is another problem. Apart from low economic status or gastrointestinal disorders, there are also other factors contributing to malnutrition. Hyper-metabolic status in β-thalassemia patients and difficulty in managing a sufficiently balanced diet are important. Reducing the consumption of food containing iron will reduce the intake of adequate calories. The recommendation for tea consumption to reduce iron absorption leads to a reduction in milk consumption, thereby increasing the essential nutrients for normal growth, such as protein, calcium, and zinc decreases [18].
Other contributing factors in growth retardation are pre-transfusion of hemoglobin level [19], lack of folic acid [16], frequent use of chelating drugs [20], emotional factors [8], low socioeconomic level [21], autoimmune factors [22], and genotype of thalassemia [7].
Regarding chelating agents, desferrioxamine is a chelator agent that is intensive and or premature use (for example, before 3 years of age and >50 mg/kg/day), and may have bone complications [23]. Taking these agents in patients with hyper transfusion is necessary because desferrioxamine binds to iron and removes excess iron from the body; however, it may cause metaphyseal long-bone dysplasia, spinal changes, and growth retardation. Desferrioxamine also prevents the synthesis of DNA and cell proliferation, collagen formation, and degradation of copper, zinc or calcium in the bone matrix. The lack of essential micronutrients reduces alkaline phosphatase activity, and decreases spinal height, thereby leading to truncal shortening, even in the presence of normal stature [24]. Body disproportion between the upper and lower body segments (the former is shorter than normal and the latter is normal) is a common feature of short stature in thalassemia patients. Male sex is suggested as an independent risk factor [19]. However, several studies argue otherwise [25-27].
The exact age of growth retardation in β-thalassemia patients does not have the same depth of research for its correlated results. According to Saxena, growth retardation occurs at the age of 8 years [21]. Therefore, most children with thalassemia have normal growth during the childhood period; however, Soliman et al. claimed that the process of growth development declines at the age of 4 [16]. Nokeaingtong et al. reported that in children with thalassemia, the mean height Z-score is reduced by 0.19 SD yearly from 5 to 14 years of age [19]. Another study that was performed in 1973 showed that short stature in boys begins at the age of 8 years and at 11 years old becomes visible. This process in girls begins at 10 years of age and becomes visible at 12 years [21].
Growth development occurs in two stages. The first occurs during the childhood period which depends on growth hormones and the second stage is during the puberty period which depends both on growth and sex hormone secretion [1, 16, 21]. In children with β-thalassemia, growth retardation occurs in both stages of childhood and puberty. However, there is a study which claimed that growth retardation in children with β-thalassemia establishes within three phases. The first phase starts in children with the age of less than 5 years because of ineffective erythropoiesis and anemia. The second phase happens between the ages of 5 to 9 years due to iron overload which affects the pituitary gland and the last phase occurs in the pre-pubertal period [18].
As different cut-off points for growth retardation in thalassemia patients are reported and short stature is more treatable in early periods, it is desirable to find a careful cut-off age for its incidence and help patients before it is too late. In this study, we tried to find such a cut-off point via statistical analysis.
Methods
Study protocol
In this cross-sectional study, data from 803 β-thalassemia major patients were analyzed. Information was extracted from questionnaires from a bone mineral densitometry (BMD) department of a referral center from around the country. The location was the Special Medical Center of Charity Foundation for Special Diseases. The questionnaires were completed during an in-person interview by a BMD operator during 2003-2010. The exclusion criteria were being a current user or chronic user of systemic steroids. Also, patients who reported a major risk factor for osteoporosis were omitted from this study. The inclusion criteria referred to the BMD department to measure height and weight carefully and with one device.
Height and weight were measured by one device as all of the BMD scans were done by one DXA machine (Norland XR-46 densitometer). A medical history was obtained about any drug or disease that positively or negatively affects BMD.
Short stature in children was defined by a height percentile of less than 3%. Hypothyroidism was defined as the current use of levothyroxine. A history of diabetes was based on the suggestion of patients.
Low bone mass was considered as Z scores ≤−2 BMD relative to age and sex-specific norms.
Deformities were reported by the BMD operator. The operator reported it in the spine or femur if deformity remained after a good positioning of patients and did not disappear after careful repositioning.
As diabetes and hypothyroidism (levothyroxine use is regarded as a surrogate of hypothyroidism), are a minor risk factor, patients with a medical history of diabetes and levothyroxine use were not omitted. On the other hand, these two diseases are known disorders by people with medications that increase the accuracy of data received from patients.
The procedures were approved by the Ethics Committee of the Endocrinology and Metabolism Research Institute of Tehran University of Medical Sciences (EMRI of TUM) and performed according to the ethical standards in the 1964 Declaration of Helsinki and its later amendments [28]. The data used for this project were obtained from routine and scientific investigations of thalassemia patients. Moreover, considering the confidentiality of this information, it does not seem to be harmful to the patients. Also, considering that the implementation of this project helps to check the health and help the health of other thalassemia patients, it seems that conducting this study without the consent letter of the patients does not create an ethical problem.
Statistical analysis
SPSS software, version 16 used for statistical analysis. Continuous variables are summarized as Means±SD and ranges. Categorical variables are provided as simple percentages. The independent samples t-test procedure was used for the comparison of means for the two groups of cases. To assess the association between dichotomous variables we used the crosstab. Statistical significance was set at 0.05.
We used an odd ratio (OR) calculation to find the cut-off age for the incidence of short stature. An odds ratio is a relative measure of effect, which allows the comparison of the intervention group of a study relative to the comparison or placebo group. Accordingly, if the outcome is the same in both groups, the ratio will be 1, which implies there is no difference between the two arms of the study. OR calculation usually contains a confidence interval (CI) that if it crosses 1, is non-significant. On the other hand, if the CI does not cross 1, it is significant. This means that one arm of the study (for example, being at an age and higher), significantly (with a 5% CI) changes the risk of an outcome (e.g. diabetes in thalassemic patients). The primary independent parameter (for example, age) must have a significant correlation with the dependent parameter (being diabetic). We used such analysis to find the cut-off age for the incidence of endocrine complications in thalassemia patients. We tried to find that being at what age (and older) significantly raises the risk of any complication that was found significantly related to age (at first, we tested this correlation).
Results
Participants’ characteristics and the prevalence of disorders
The mean age of patients was 20±7 years old and female to male ratio was 420/383. Children (<20 years of age) formed 47% of the participants. The mean ages of males and females were 20.1 and 19.9 years, respectively (no significant difference, P=0.693). Short stature was found in 32% of patients and the youngest short stature patient was 6 years old. Diabetes and hypothyroidism were found in 3.3% and 2% of cases, respectively. The prevalence of low BMD of the neck and spine was almost equal and found in 30% of the participants.
Associations
Significant correlations were found between age, sex, diabetes, and short stature (P≤0.001, 0.042 and <0.001, respectively). Male sex was a risk factor for short stature.
Age has a significant correlation with short stature and it is a continuous parameter. An OR calculation was used to find which age significantly increases the chance of being short and considered it as the cut-off age for short stature. Being 10 and 11 years old and above increased the risk of short stature, 28.5 and 21.4 times, respectively compared to younger patients (both P<0.001). On the other hand, after 7 years of age up to 18 years of age, similar to 10 years of age and 11 years of age, the risk of short stature was increased. Also, being 27 years old and 30 years old and above increases the risk of short stature by 1.4 and 1.7 times, respectively, compared to younger patients (P=0.042 and <0.018, respectively). In Table 1, significant increases in the risk of short stature according to age levels are listed. In this table, we also showed the results for men and women, separately.
Discussion
This study found the prevalence of short stature as high as 32%. Meanwhile, the prevalence of short stature was reported previously at 31% to 55% [2]. Accordingly, the prevalence of short stature in our patients is among the lowest rates. This can be considered the result of the implementation of an active health network service for thalassemic patients in Iran. The thalassemia prevention program was established in 1995. It is extended all over the country (it is based in 64 medical universities and faculties). Almost all thalassemia patients are supported by these centers. It is reported that the mean hemoglobin of patients on the day of transfusion is almost 8.0 g/dL. This can be translated to a relatively good distribution of blood supply for them. Besides, for appropriate management of iron overload, iron chelation agents that are internally manufactured, are free for all thalassemic patients.
In previous studies, 2 and 3 phases of growth retardation were reported in thalassemia patients. Our findings are closer to authors that suggest 3 phases or cut-offs for growth retardation because we found ages of 7 years, 11 years and 18 years as cut-off points. However, according to our findings, growth retardation in thalassemic patients is a continuous phenomenon. Maybe the reason behind this is the incident of deformities due to micro compression fractures of vertebrae, a continuous problem. In this cross-section, we found 4% deformity in the spine 1% deformity in the femur, and 30% of low BMD. However, the results may be different from one ethnicity to another [29].
Male to female difference in short stature prevalence is significant and males have more short stature than females. This is in line with the study by Nokeaingtong et al. They found that children’s height-for-age Z-scores decreased up to the age of 14 (from the age of 5). But at 14 years of age, females showed a growth spurt, whereas growth in male children continued to decline. Maybe females show a better response to hormones or less detrimental reactions to iron overload. Even some investigators suggested they have lesser sensitivity to chronic oxidative stress [19]. But there are studies in disagreement [25-27]. In a study by Altincik al., short stature was present in 42% and in men and women, the prevalence was almost the same [25]. In another population of 40 females and 40 males of β thalassemia patients, the number of short-stature patients was almost the same (33 male, 32 female) [26]. Rathaur et al. found that 14/24 females and 32/46 males were short, but no significant association was found between sex and short stature [27].
Diabetes was found to have a direct correlation with short stature while hypothyroidism did not correlate. We found no scientific reason for these correlations but the late peak of increased risk of short stature at 27 and 30 years of age may be due to an increase in diabetes risk at these ages in thalassemic patients [30].
Conclusion
We suggest that awareness about short stature and monitoring for it in thalassemic patients should be done in their childhood. Given that being 7 years old increases the risk of short stature, we suggest the start of monitoring as early as 5 years of age. We hope that with these studies we improve thalassemic patients’ management while enabling a better quality of life for them.
Study limitations
This study faced some limitations. The lack of information about hypogonadism and delayed puberty in patients was a limitation (data about females is consistent but no question about males’ age of puberty or hypogonadism was asked, so we ignored all data about hypogonadism and delayed puberty). Also, a DXA scan is not the gold standard for diagnosis of spinal or femoral deformity, but if we do not find any deformity in the spinal region in a DXA scan, it is confirmatory. Another limitation of our study was that no data about hormonal lab tests existed in our questionnaire. Although there are some limitations, this study has strong aspects, such as a high number of patients and measurement in a referral center, by one device, and one operator (95% measurement done by one operator). Another novelty of this study is using statistical tools for finding cut-off points of short stature, that were not used or not referred to in previous studies. The statistical test that we used for defining cut-off points in this study is also important, because by using it, we could determine the OR between short stature and age cut-off points that we determined.
Ethical Considerations
Compliance with ethical guidelines
The procedures were approved by the Ethics Committee of the Endocrinology and Metabolism Research Institute of Tehran University of Medical Sciences (EMRI of TUM) (Code: EC-00355) and performed according to the ethical standards in the 1964 Declaration of Helsinki and its later amendments
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Study design: Fariba Mohseni, Mohammad-Reza Mohajeri-Tehrani, Bagher Larijani and Zohreh Hamidi; Study conduct: Zohreh Hamidi; Data collection: Najmeh Darvishian, Faezeh Arab, Sedigheh Salemkar and Zohreh Hamidi; Data analysis: Zohreh Hamidi; Data interpretation: Mohammad Reza Mohajeri-Tehrani, Fariba Mohseni and Zohreh Hamidi; Drafting the manuscript: Zohreh Hamidi; Revising manuscript content: Fariba Mohseni and Mohammad-Reza Mohajeri-Tehrani; Final approval: Mohammad-Reza Mohajeri-Tehrani.
Conflicts of interest
The authors declared no conflict of interest.
References
- Lampl M, Schoen M. How long bones grow children: Mechanistic paths to variation in human height growth. Am J Hum Biol. 2017; 29(2). [DOI:10.1002/ajhb.22983] [PMID]
- Mohseni F, Mohajeri-Tehrani MR, Larijani B, Hamidi Z. Relation between BMD and biochemical, transfusion and endocrinological parameters in pediatric thalassemic patients. Arch Osteoporos. 2014;9:174. [DOI:10.1007/s11657-014-0174-3] [PMID]
- Najafipour F, Aliasgarzadeh A, Aghamohamadzadeh N, Bahrami A, Mobasri M, Niafar M, et al. A cross-sectional study of metabolic and endocrine complications in beta-Thalassemia major. Ann Saudi Med. 2008; 28(5):361-6. [DOI:10.5144/0256-4947.2008.361] [PMID]
- Sharma R, Seth A, Chandra J, Gohain S, Kapoor S, Singh P, Pemde H. Endocrinopathies in adolescents with thalassaemia major receiving oral iron chelation therapy. Paediatr Int Child Health. 2016; 36(1):22-7. [DOI:10.1179/2046905514Y.0000000160] [PMID]
- Ong CK, Lim SL, Tan WC, Ong EE, Goh AS. Endocrine complications in transfusion dependent Thalassaemia in Penang Hospital. Med J Malaysia. 2008; 63(2):109-2. [Link]
- Fahim FM, Saad K, Askar EA, Eldin EN, Thabet AF. Growth parameters and vitamin D status in children with Thalassemia major in Upper Egypt. Int J Hematol Oncol Stem Cell Res. 2013; 7(4):10-4. [PMID]
- Yin XL, Wu ZK, He YY, Zhou TH, Zhou YL, Zhang XH. Treatment and complications of Thalassemia major in Guangxi, Southern China. Pediatr Blood Cancer. 2011; 57(7):1174-8. [DOI:10.1002/pbc.23101] [PMID]
- Hamidi Z. Endocrine Disorders in thalassemia major patients: A review. Kuwait Med J. 2016; 48 (1):4-11. [Link]
- Merchant RH, Shirodkar A, Ahmed J. Evaluation of growth, puberty and endocrine dysfunctions in relation to iron overload in multi transfused Indian Thalassemia patients. Indian J Pediatr. 2011; 78(6):679-83. [DOI:10.1007/s12098-010-0351-3] [PMID]
- Belhoul KM, Bakir ML, Saned MS, Kadhim AM, Musallam KM, Taher AT. Serum ferritin levels and endocrinopathy in medically treated patients with β Thalassemia major. Ann Hematol. 2012; 91(7):1107-14. [DOI:10.1007/s00277-012-1412-7] [PMID]
- Bazi A, Sharifi-Rad J, Rostami D, Sargazi-Aval O, Safa A. Diabetes mellitus in Thalassaemia major patients: A report from the Southeast of Iran. J Clin Diagn Res. 2017; 11(5):BC01-4. [DOI:10.7860/JCDR/2017/24762.9806] [PMID]
- Mula-Abed WA, Al Hashmi H, Al Muslahi M, Al Muslahi H, Al Lamki M. Prevalence of endocrinopathies in patients with Beta-thalassaemia major - A cross-sectional study in oman. Oman Med J. 2008; 23(4):257-62. [PMID]
- De Sanctis V, Soliman AT, Elsefdy H, Soliman N, Bedair E, Fiscina B, et al. Bone disease in β Thalassemia patients: Past, present and future perspectives. Metabolism. 2018; 80:66-79. [DOI:10.1016/j.metabol.2017.09.012] [PMID]
- Ho SW, Kwek EB. Multiple pathological fractures secondary to endocrinopathy from Thalassaemia. Ann Acad Med Singap. 2016; 45(7):318-21. [DOI:10.47102/annals-acadmedsg.V45N7p318] [PMID]
- Wood JC, Noetzl L, Hyderi A, Joukar M, Coates T, Mittelman S. Predicting pituitary iron and endocrine dysfunction. Ann N Y Acad Sci. 2010; 1202:123-8. [DOI:10.1111/j.1749-6632.2010.05545.x] [PMID]
- Soliman AT, Khalafallah H, Ashour R. Growth and factors affecting it in Thalassemia major. Hemoglobin. 2009; 33 Suppl 1:S116-26. [DOI:10.3109/03630260903347781] [PMID]
- Wassner AJ. Pediatric hypothyroidism: Diagnosis and treatment. Paediatr Drugs. 2017; 19(4):291-301. [DOI:10.1007/s40272-017-0238-0] [PMID]
- Fung EB. The importance of nutrition for health in patients with transfusion-dependent Thalassemia. Ann N Y Acad Sci. 2016; 1368(1):40-8. [DOI:10.1111/nyas.13003] [PMID]
- Nokeaingtong K, Charoenkwan P, Silvilairat S, Saekho S, Pongprot Y, Dejkhamron P. A longitudinal study of growth and relation with anemia and iron overload in pediatric patients with transfusion-dependent Thalassemia. J Pediatr Hematol Oncol. 2016; 38(6):457-62. [DOI:10.1097/MPH.0000000000000625] [PMID]
- Cao A, Galanello R. Beta-Thalassemia. Genet Med. 2010; 12(2):61-76. [DOI:10.1097/GIM.0b013e3181cd68ed] [PMID]
- Saxena A. Growth retardation in Thalassemia major patients. Int J Hum Genet. 2003; 3(4):237-46. [DOI:10.1080/09723757.2003.11885858]
- Borgna-Pignatti C, De Stefano P, Zonta L, Vullo C, De Sanctis V, Melevendi C, et al. Growth and sexual maturation in Thalassemia major. J Pediatr. 1985; 106(1):150-5. [DOI:10.1016/S0022-3476(85)80488-1] [PMID]
- Tyler PA, Madani G, Chaudhuri R, Wilson LF, Dick EA. The radiological appearances of Thalassaemia. Clin Radiol. 2006; 61(1):40-52. [DOI:10.1016/j.crad.2005.07.006] [PMID]
- Kyriakou A, Skordis N. Thalassaemia and aberrations of growth and puberty. Mediterr J Hematol Infect Dis. 2009; 1(1):e2009003. [PMID]
- Altincik A, Akin M. Prevalence of endocrinopathies in Turkish children with β-Thalassemia major: A single-center study. J Pediatr Hematol Oncol. 2016; 38(5):389-93. [DOI:10.1097/MPH.0000000000000573] [PMID]
- Elizabeth M, Fadlyana E, Reniarti L, Faisal F, Sukandar H, Rusmil K. Serum IGF-1 and short stature in adolescents with β-Thalassemia major. Paediatrica Indonesiana. 2018; 58(4):151-8. [DOI:10.14238/pi58.4.2018.151-8]
- Rathaur VK, Imran A, Pathania M. Growth pattern in thalassemic children and their correlation with serum ferritin. J Family Med Prim Care. 2020; 9(2):1166-9. [DOI:10.4103/jfmpc.jfmpc_951_19] [PMID]
- Declaration of Helsinki (DoH). Medical research involving human subjects. Helsinki: DoH; 2023. [Link]
- Mohajeri-Tehrani MR, Darvishian N, Arab F, Salemkar S, Mohseni F, Larijani B, et al. The role of using different reference population in the prevalence of low BMD in the Thalassemia patients. J Diabetes Metab Disord. 2019; 19(1):431-5. [DOI:10.1007/s40200-019-00455-6] [PMID]
- Mohajeri-Tehrani MR, Alemzadeh SA, Abbaszadeh Marzbali F, Nasserisina S, Hosnan F, Naghghash A, et al. Some determinants of endocrine and bone disorders in Thalassemia major patients. Iran J Public Health. 2023; 53(2):433-42. [DOI:10.18502/ijph.v53i2.14928]
Type of Study: Research Article |
Subject:
Endocrinology
Received: 2022/10/18 | Accepted: 2023/09/23 | Published: 2024/01/1
Received: 2022/10/18 | Accepted: 2023/09/23 | Published: 2024/01/1
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
![]()
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC),
which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
